Understanding Cellular Dynamics with RNA Velocity
Briefly speaking, RNA velocity is a single cell RNA-seq computational method to predict future gene expression states based on the ratio of spliced and unspliced mRNA transcripts.
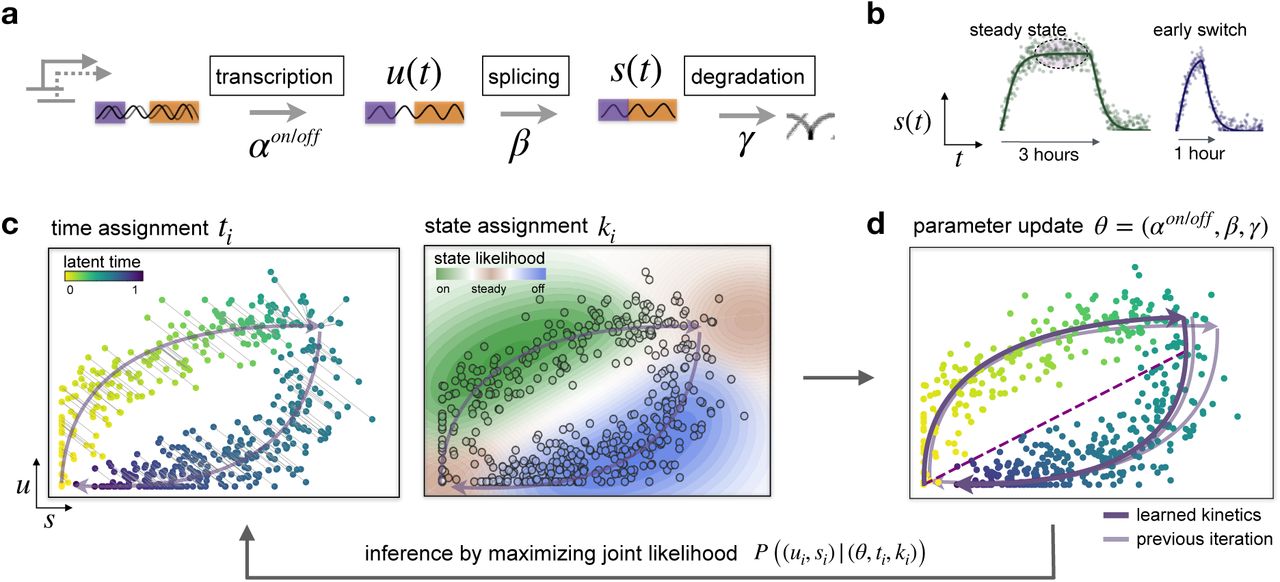
The original RNA velocity model, Velocyto, assumes a constant rate of transcription for each gene solving a system of linear differential equations. Later, scVelo was developed to improve the original implementation. It uses a dynamic model with a shared latent time across all cells and fits gene-specific transcription, splicing, and degradation parameters by likelihood-based expectation maximization
See the following animation by the scVelo team to better understand the relationship of the spliced vs. unspliced phase portrait with the abundance of pre-mRNA (unspliced) and mature mRNA (spliced)
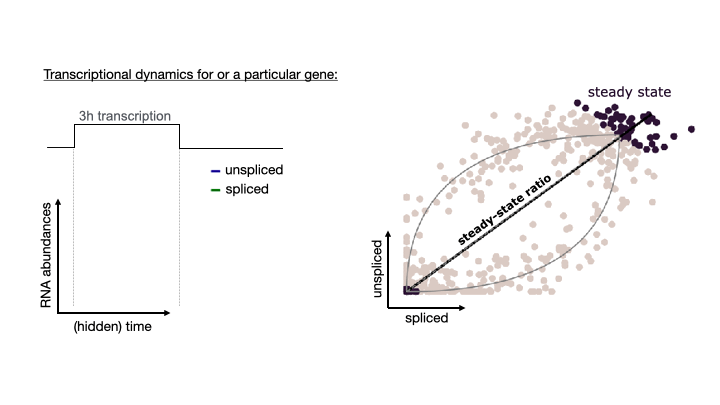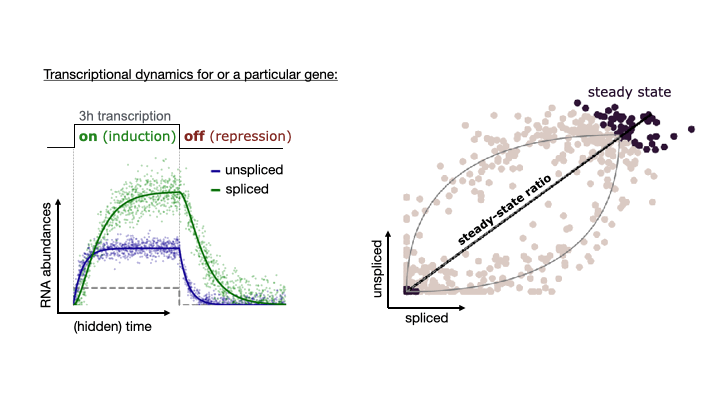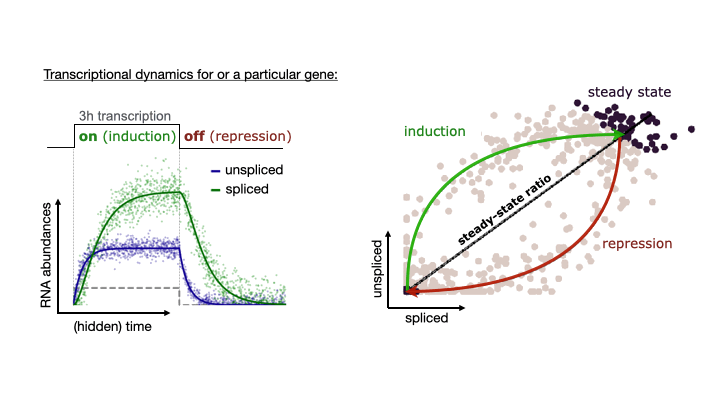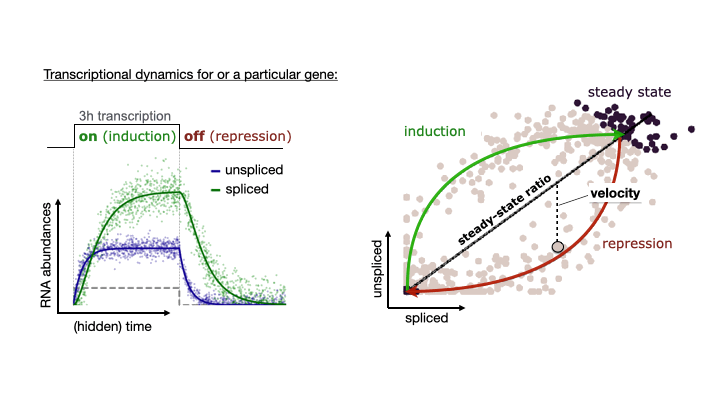
For this post, I will use scVelo to estimate RNA velocity using the dynamical model of the same datasets used in my Pseudotime Trajectories with Slingshot post
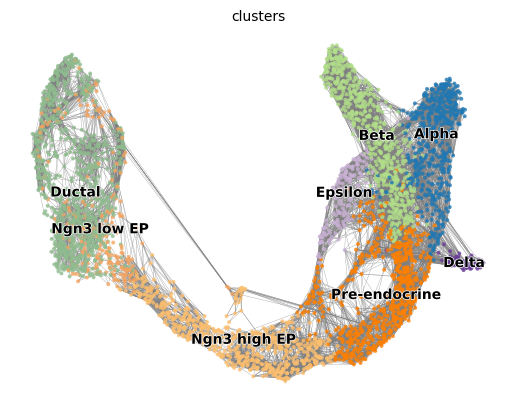
Pancreas dataset
As in the Slingshot post, we use the dataset from Bastidas-Ponce et al. (2018) downloaded from scVelo github (https://github.com/theislab/scvelo_notebooks/tree/master/data/Pancreas/endocrinogenesis_day15.h5ad).
we start importing the required python packages
1import scanpy as sc
2import scvelo as scv
Next read the h5ad file as follows:
1adata = sc.read('endocrinogenesis_day15.h5ad')
and check the contents
1adata
AnnData object with n_obs × n_vars = 3696 × 27998
obs: 'clusters_coarse', 'clusters', 'S_score', 'G2M_score'
var: 'highly_variable_genes'
uns: 'clusters_coarse_colors', 'clusters_colors', 'day_colors', 'neighbors', 'pca'
obsm: 'X_pca', 'X_umap'
layers: 'spliced', 'unspliced'
obsp: 'distances', 'connectivities'
We start with standard preprocessing of the data
1scv.pp.filter_genes(adata, min_shared_counts=20)
2scv.pp.normalize_per_cell(adata)
3scv.pp.filter_genes_dispersion(adata, n_top_genes=2000)
4sc.pp.log1p(adata)
Filtered out 20801 genes that are detected 20 counts (shared).
Normalized count data: X, spliced, unspliced.
Extracted 2000 highly variable genes.
Next calculate first and second order moments
1scv.pp.moments(adata, n_pcs=30, n_neighbors=30)
computing neighbors
finished (0:00:05) --> added
'distances' and 'connectivities', weighted adjacency matrices (adata.obsp)
computing moments based on connectivities
finished (0:00:00) --> added
'Ms' and 'Mu', moments of un/spliced abundances (adata.layers)
We now run the dynamical model to infer the complete transcriptional dynamics underlying splicing kinetics. This is achieved using a likelihood-based expectation-maximization framework, which iteratively estimates both the reaction rate parameters and latent cell-specific variables. These are the transcriptional state and each cell’s internal latent time. The goal is to reconstruct the unspliced/spliced phase trajectory for each gene.
1scv.tl.recover_dynamics(adata)
recovering dynamics (using 1/8 cores)
WARNING: Unable to create progress bar. Consider installing `tqdm` as `pip install tqdm` and `ipywidgets` as `pip install ipywidgets`,
or disable the progress bar using `show_progress_bar=False`.
finished (0:06:24) --> added
'fit_pars', fitted parameters for splicing dynamics (adata.var)
Now we can estimate RNA velocity using the dynamical model, which are stored in adata.layers.
1scv.tl.velocity(adata, mode='dynamical')
computing velocities
finished (0:00:02) --> added
'velocity', velocity vectors for each individual cell (adata.layers)
The combination of velocities across genes can then be used to estimate the future state of an individual cell. In order to project the velocities into a lower-dimensional embedding, transition probabilities of cell-to-cell transitions are estimated. That is, for each velocity vector we find the likely cell transitions that are accordance with that direction. The transition probabilities are computed using cosine correlation between the potential cell-to-cell transitions and the velocity vector, and are stored in a matrix denoted as velocity graph.
— scVelo tutorial
1scv.tl.velocity_graph(adata)
computing velocity graph (using 1/8 cores)
finished (0:00:05) --> added
'velocity_graph', sparse matrix with cosine correlations (adata.uns)
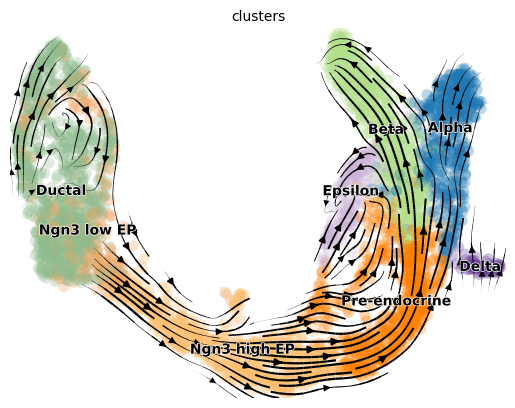
Project the velocities onto UMAP as streamlines
1scv.pl.velocity_embedding_stream(adata, basis='umap')
computing velocity embedding
finished (0:00:00) --> added
'velocity_umap', embedded velocity vectors (adata.obsm)
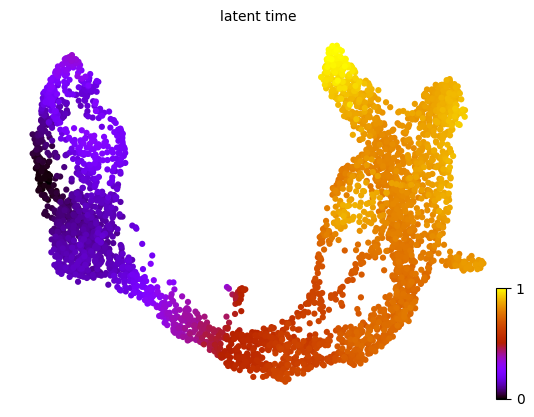
Latent time
The dynamical model infers the latent time of the underlying cellular processes, capturing each cell’s internal clock. This latent time reflects an estimate of the actual progression experienced by cells during differentiation, derived solely from the transcriptomic data.
1scv.tl.latent_time(adata)
2scv.pl.scatter(adata, color='latent_time', color_map='gnuplot', size=80)
computing terminal states
identified 2 regions of root cells and 1 region of end points .
finished (0:00:00) --> added
'root_cells', root cells of Markov diffusion process (adata.obs)
'end_points', end points of Markov diffusion process (adata.obs)
computing latent time using root_cells as prior
finished (0:00:00) --> added
'latent_time', shared time (adata.obs)
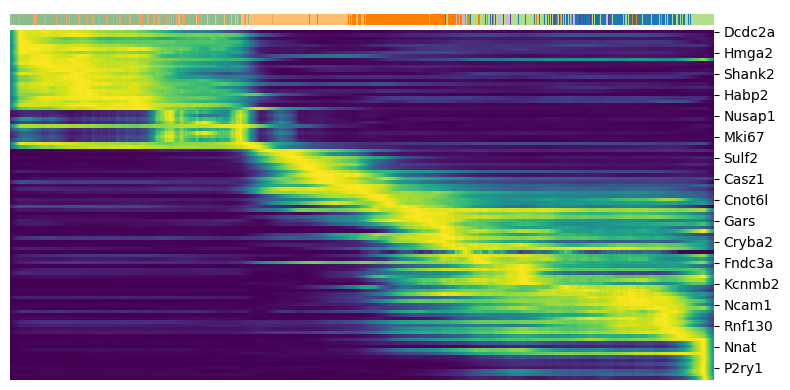
Driver genes are selected as those with high likelihoods in the dynamic model. The likelihood of a gene is defined as the probability of observing the data given the model parameters. The higher the likelihood, the better the model fits the data. Genes with high likelihoods are more likely to be involved in the underlying biological process.
1top_genes = adata.var['fit_likelihood'].sort_values(ascending=False).index[:100]
2scv.pl.heatmap(adata, var_names=top_genes, sortby='latent_time', col_color='clusters', n_convolve=100)
Here we check a few of the top likelihood genes using phase portraits.
1top_genes = adata.var['fit_likelihood'].sort_values(ascending=False).index
2scv.pl.scatter(adata, basis=top_genes[:6], ncols=3, frameon=False)
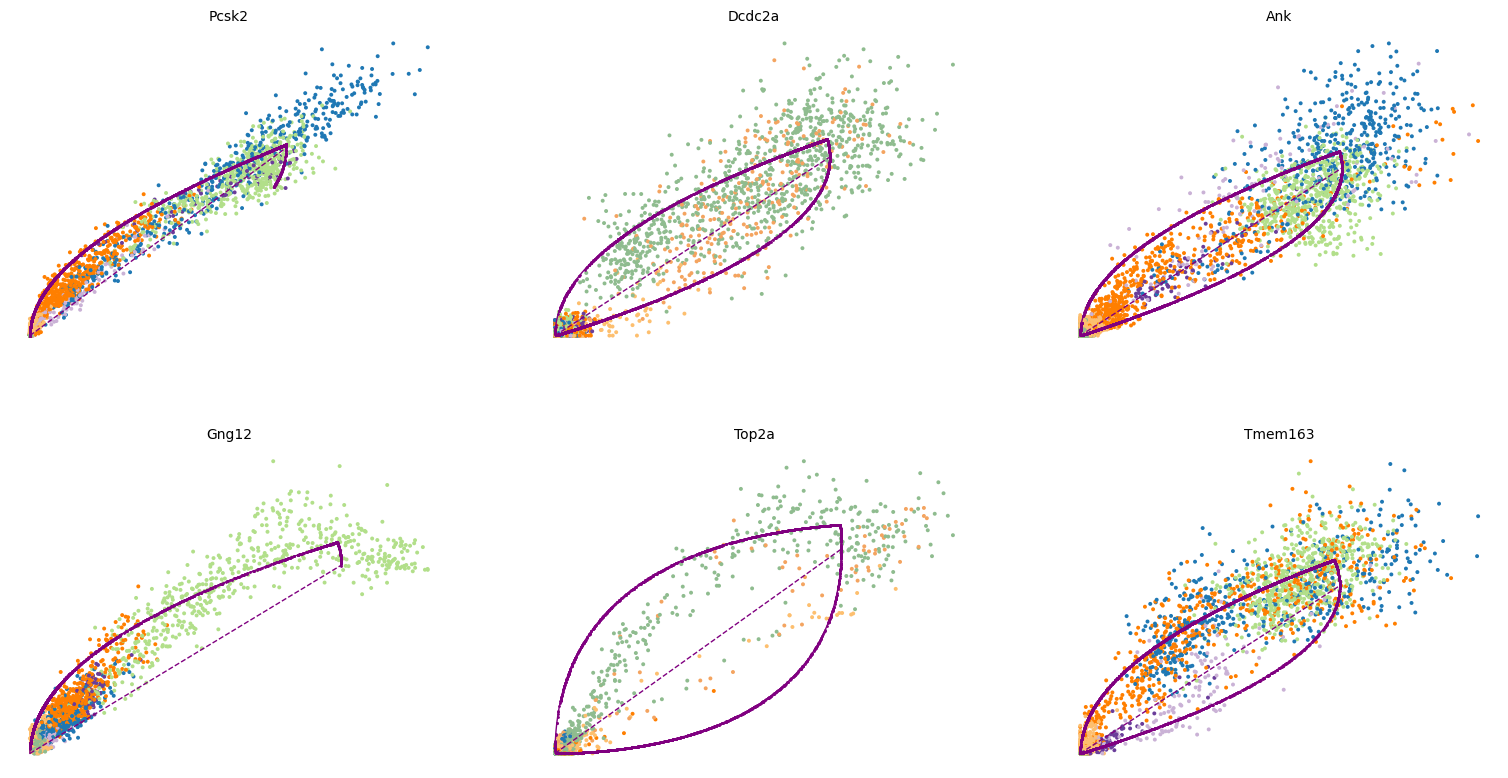
and their expression along the calculated latent time
1scv.pl.scatter(adata, x='latent_time', y=top_genes.values[:6], frameon=False)
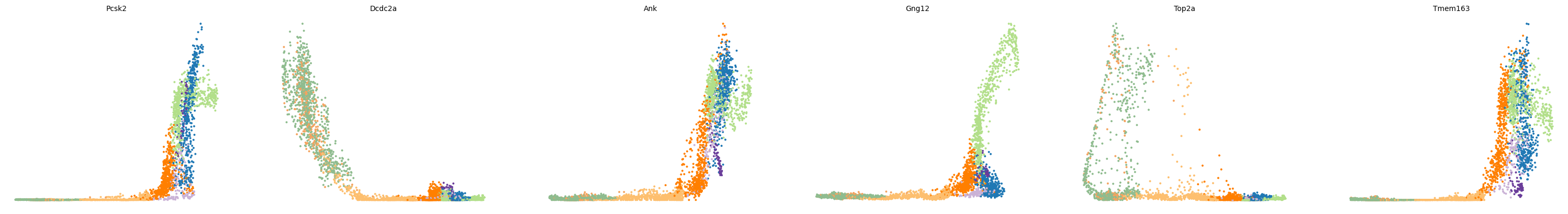
We can plot the velocity graph to visualize all velocity-inferred cell connections/transitions.
1scv.pl.velocity_graph(adata, threshold=.1)
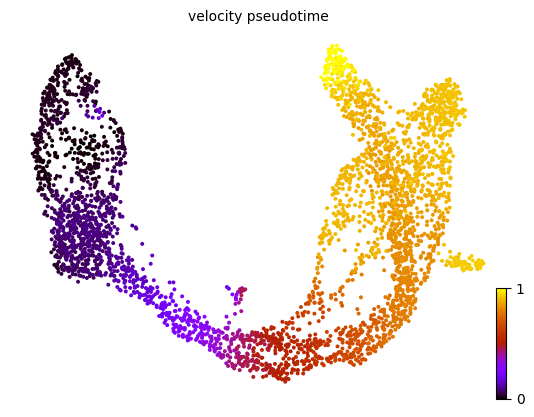
based on the velocity graph, a velocity pseudotime can be computed.
1scv.tl.velocity_pseudotime(adata)
2scv.pl.scatter(adata, color='velocity_pseudotime', cmap='gnuplot')
save the object
1adata.write('pancreas.withVelocity.h5ad')
Spermatogenesis Data
Again, we use the same dataset as in the Slingshot post from Hermann et al. (2018), downloaded from the scRNAseq package and coverted to h5ad format using the zellkonverter package in R as follows
1library(zellkonverter)
2writeH5AD(sce.sling, file = 'spermatogenesis.slingshot.h5ad', verbose = TRUE)
So we can read the h5ad file as follows in python
1adata = sc.read('spermatogenesis.slingshot.h5ad')
and check the contents
1adata
AnnData object with n_obs × n_vars = 1816 × 29826
obs: 'celltype', 'sum', 'detected', 'subsets_Mt_sum', 'subsets_Mt_detected', 'subsets_Mt_percent', 'total', 'sizeFactor', 'slingPseudotime_1'
uns: '.colData', 'X_name'
obsm: 'PCA', 'UMAP', 'UMAP_3D'
layers: 'spliced', 'unspliced'
the pl.embedding function expects a numpy array but the conversor generated a pandas df
1adata.obsm["UMAP"]
| UMAP1 | UMAP2 | |
|---|---|---|
| CCCATACTCCGAAGAG | 8.908866 | 5.790795 |
| AATCCAGTCATCTGCC | 9.038168 | 5.535748 |
| GACTGCGGTTTGACTG | 9.124887 | 5.500014 |
| ACACCAATCTTCGGTC | 9.187134 | 5.613092 |
| TGACAACAGGACAGAA | 2.905884 | -6.314294 |
| ... | ... | ... |
| TGCGTGGGTATATGGA | -9.742685 | 7.182144 |
| TTAACTCAGTTGAGAT | 6.960753 | 5.540197 |
| TAGCCGGAGGAATCGC | -9.991141 | 6.813681 |
| TCGCGTTCAAGAGTCG | -1.258456 | 4.865719 |
| CACCTTGCAGATCGGA | -1.269776 | 4.853017 |
1816 rows × 2 columns
1print(type(adata.obsm['UMAP']))
<class 'pandas.core.frame.DataFrame'>
so it must be converted to a numpy array
1adata.obsm['UMAP'] = adata.obsm['UMAP'].to_numpy()
Now we can plot the UMAP embedding
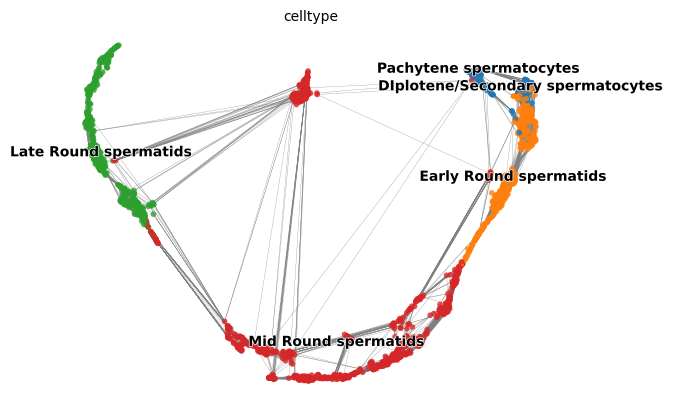
1sc.pl.embedding(adata, basis = 'UMAP', color = 'celltype' )
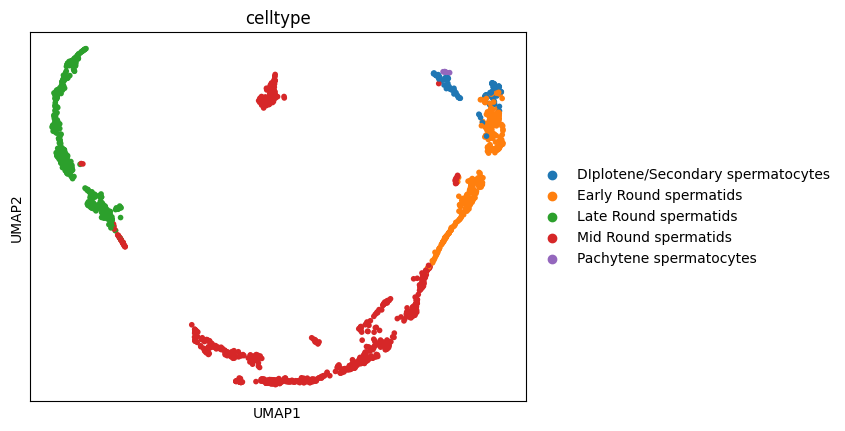
Since the data was already preprocessed in the Slingshot post we can directly calculate the first and second order moments
1scv.pp.moments(adata, n_pcs=30, n_neighbors=30)
computing neighbors
finished (0:00:00) --> added
'distances' and 'connectivities', weighted adjacency matrices (adata.obsp)
computing moments based on connectivities
finished (0:00:02) --> added
'Ms' and 'Mu', moments of un/spliced abundances (adata.layers)
it recomputes PCA to perform the moments calculations since it was not stored with the expected key.
1adata
AnnData object with n_obs × n_vars = 1816 × 29826
obs: 'celltype', 'sum', 'detected', 'subsets_Mt_sum', 'subsets_Mt_detected', 'subsets_Mt_percent', 'total', 'sizeFactor', 'slingPseudotime_1'
uns: '.colData', 'X_name', 'celltype_colors', 'pca', 'neighbors'
obsm: 'PCA', 'UMAP', 'UMAP_3D', 'X_pca'
varm: 'PCs'
layers: 'spliced', 'unspliced', 'Ms', 'Mu'
obsp: 'distances', 'connectivities'
run dynamical model
1scv.tl.recover_dynamics(adata)
recovering dynamics (using 1/8 cores)
finished (0:24:11) --> added
'fit_pars', fitted parameters for splicing dynamics (adata.var)
and Estimate RNA velocity
1scv.tl.velocity(adata, mode='dynamical')
2scv.tl.velocity_graph(adata)
computing velocities
finished (0:00:08) --> added
'velocity', velocity vectors for each individual cell (adata.layers)
computing velocity graph (using 1/8 cores)
finished (0:00:14) --> added
'velocity_graph', sparse matrix with cosine correlations (adata.uns)
project the velocities onto UMAP as streamlines
1scv.pl.velocity_embedding_stream(adata, basis='UMAP', color = 'celltype' )
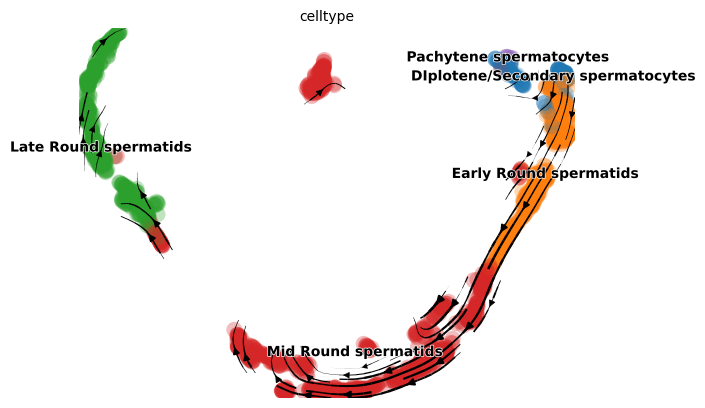
calculate latent time
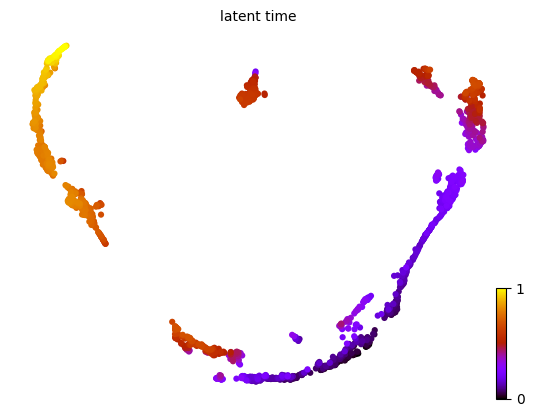
1scv.tl.latent_time(adata)
2scv.pl.scatter(adata, basis='UMAP', color='latent_time', color_map='gnuplot', size=80)
computing terminal states
identified 1 region of root cells and 1 region of end points .
finished (0:00:00) --> added
'root_cells', root cells of Markov diffusion process (adata.obs)
'end_points', end points of Markov diffusion process (adata.obs)
computing latent time using root_cells as prior
finished (0:00:02) --> added
'latent_time', shared time (adata.obs)
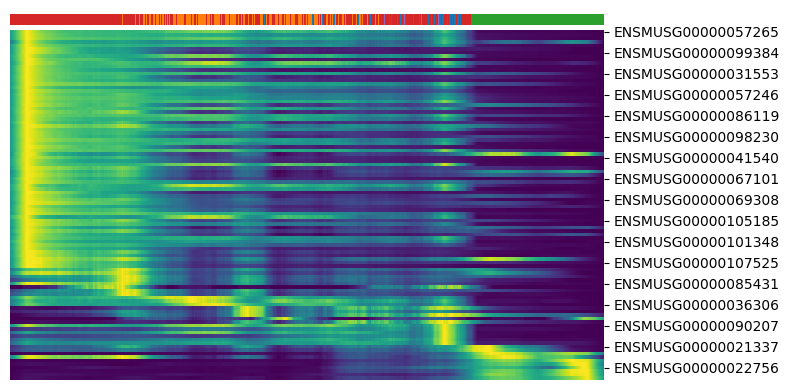
get the driver genes and plot the heatmap
1top_genes = adata.var['fit_likelihood'].sort_values(ascending=False).index[:100]
2scv.pl.heatmap(adata, var_names=top_genes, sortby='latent_time', col_color='celltype', n_convolve=100)
/home/alfonso/.virtualenvs/scvelo/lib/python3.10/site-packages/scvelo/plotting/utils.py:68: DeprecationWarning: is_categorical_dtype is deprecated and will be removed in a future version. Use isinstance(dtype, pd.CategoricalDtype) instead
return isinstance(c, str) and c in data.obs.keys() and cat(data.obs[c])
and scatter plots
1top_genes = adata.var['fit_likelihood'].sort_values(ascending=False).index
2scv.pl.scatter(adata, basis=top_genes[:6], ncols=3, frameon=False, color='celltype')
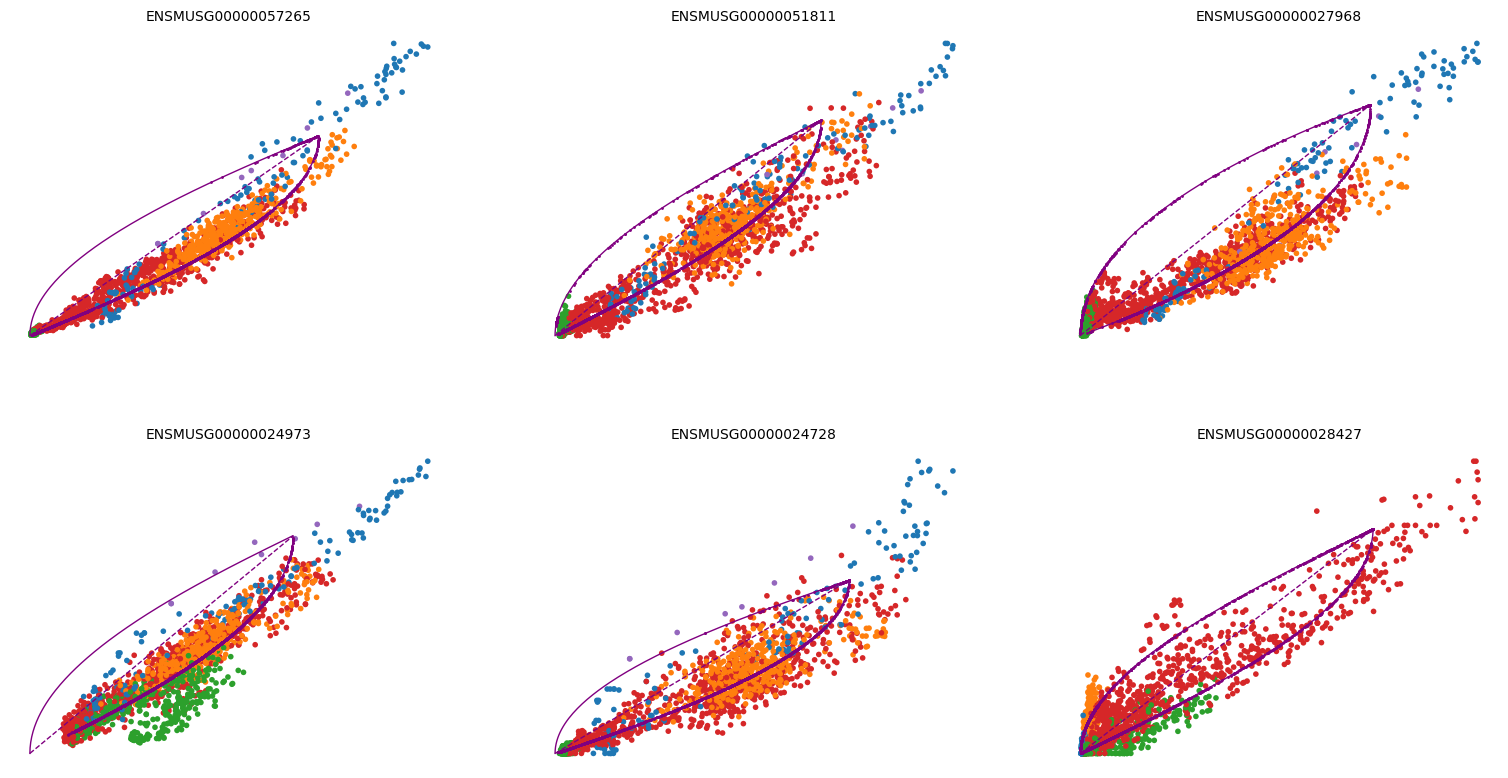
1scv.pl.scatter(adata, x='latent_time', y=top_genes.values[:6], frameon=False, color='celltype')

and Estimate RNA velocity
1scv.pl.velocity_graph(adata, basis = 'UMAP', threshold=.1, color='celltype')
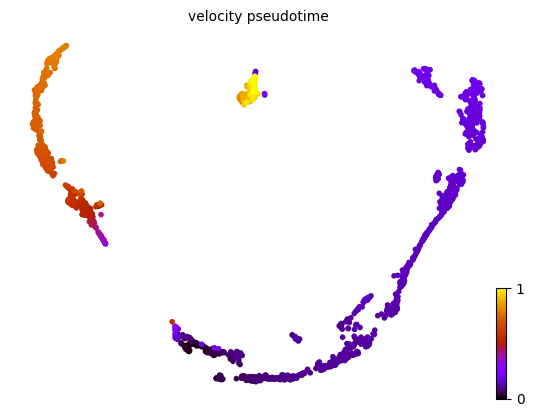
1scv.tl.velocity_pseudotime(adata)
2scv.pl.scatter(adata, basis = 'UMAP', color='velocity_pseudotime', cmap='gnuplot')
save the object as follows
1adata.write('spermatogenesis.withVelocity.h5ad')
Conclusions
- The dynamic model of scVelo is able to decipher distinct kinetic behaviors (e.g., transcriptional activity, splicing rates, differentiation timing) to describe the underlying differentiation processes.
- Selecting genes with high likelihood successfully identifies dynamically changing genes
Take away
- scVelo calculates two differential times: latent time and velocity pseudotime
- Latent time is calculated directly from the count values.
- Velocity pseudotime is a diffusion pseudotime computed based on the scVelo velocity graph and root cells obtained from the velocity-inferred transition matrix. You can see it as being analogous to the pseudotime calculated by Slingshot.
- According to the authors, latent time truly captures the underlying differentiation time.
Further reading
- La Manno, et al. RNA velocity of single cells. Nature 560, 494–498 (2018)
- Bergen, et al. Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol 38, 1408–1414 (2020)
- scVelo documentation
- RNA velocity—current challenges and future perspectives. Molecular Systems Biology. EMBO press
- “Orchestrating Single-Cell Analysis with Bioconductor” book. RNA velocity
- Single-cell best practices: RNA velocity
- A guide to trajectory inference and RNA velocity
- Challenges and Progress in RNA Velocity: Comparative Analysis Across Multiple Biological Contexts