Pseudoalignment for single cell RNA-seq with alevin-fry
The single cell RNA-seq world is dominated by alignment tools such as Cell Ranger and STARsolo, which are great but computationally expensive. Alevin-fry is a new tool that uses pseudoalignment to achieve fast and efficient quantification of single cell RNA-seq data.
In this post we will test alevin-fry using simpleaf. Simpleaf (simple alevin-fry) is a tool created to simplify the whole process in just two steps:
simpleaf indexto create an index from the reference.simpleaf quantto generate a gene count matrix by mapping the sequencing reads (FASTQ files) against the indexed reference.
For this post, I will follow this tutorial.
Download files
Download and untar 10X genomics reference into the working directory.
1cd /media/alfonso/data/alevinfry_PBMCs/reference/
2wget https://cf.10xgenomics.com/supp/cell-exp/refdata-gex-GRCh38-2024-A.tar.gz
3tar -xvf refdata-gex-GRCh38-2024-A.tar.gz
I will use the 5k Human PBMCs (Donor 3) dataset
1cd /media/alfonso/data/alevinfry_PBMCs/rawData/
2wget https://cf.10xgenomics.com/samples/cell-exp/9.0.0/5k_Human_Donor3_PBMC_3p_gem-x_Multiplex/5k_Human_Donor3_PBMC_3p_gem-x_Multiplex_fastqs.tar
3tar -xvf 5k_Human_Donor3_PBMC_3p_gem-x_Multiplex_fastqs.tar
Install simpleaf
The tutorial recommends installing simpleaf using conda. However, I will use a docker image provided by the authors. This will make our live much easier, if you do not know about docker I highly recommend you to learn and use it.
You can just use the docker run command, it will download the image if it is not yet available in your system, as follows:
1docker run --rm -it \
2 -v /media/alfonso/data/alevinfry_PBMCs:/workdir \
3 quay.io/biocontainers/simpleaf:0.19.5--ha6fb395_0 bash
With --rm we are telling Docker to remove the container once we exit it, so it does not take up space on the disk after the analysis is done.
With -v we are mounting the /media/alfonso/data/alevinfry_PBMCs directory to the /workdir directory inside the container, so we can access the local files from within the container.
With -it we are running the container in interactive mode, so we can run commands inside it as if it were a normal terminal. The last bash ask the container to launch a bash shell, so we can run commands inside it.
From now on, all commands are run inside the docker container unless otherwise stated.
Set up the variables
1export AF_SAMPLE_DIR=/workdir
2export ALEVIN_FRY_HOME="$AF_SAMPLE_DIR/af_home"
Simpleaf configuration
1simpleaf set-paths
2ulimit -n 2048
Build the Reference Index
Set variables
1REF_DIR="$AF_SAMPLE_DIR/reference/refdata-gex-GRCh38-2024-A"
2IDX_DIR="$AF_SAMPLE_DIR/human-2024-A_splici"
Create index
1simpleaf index \
2 --output $IDX_DIR \
3 --fasta $REF_DIR/fasta/genome.fa \
4 --gtf $REF_DIR/genes/genes.gtf.gz \
5 --threads 6 \
6 --work-dir $AF_SAMPLE_DIR/workdir.noindex
Quantify Gene Expression
First we get the FASTQ filenames from their directory (and subdirectory)
1FASTQ_DIR="$AF_SAMPLE_DIR/rawData/5k_Human_Donor3_PBMC_3p_gem-x_GEX_fastqs"
2
3# Define filename pattern
4reads1_pat="_R1_"
5reads2_pat="_R2_"
6
7# Obtain and sort filenames
8reads1="$(find -L ${FASTQ_DIR} -name "*$reads1_pat*" -type f | sort | awk -v OFS=, '{$1=$1;print}' | paste -sd, -)"
9reads2="$(find -L ${FASTQ_DIR} -name "*$reads2_pat*" -type f | sort | awk -v OFS=, '{$1=$1;print}' | paste -sd, -)"
Run the quantification command
1simpleaf quant \
2 --reads1 $reads1 \
3 --reads2 $reads2 \
4 --threads 4 \
5 --index $IDX_DIR/index \
6 --chemistry 10xv4-3p --resolution cr-like \
7 --unfiltered-pl --anndata-out \
8 --output $AF_SAMPLE_DIR/5k_Human_Donor3_quant
and done! The whole indexing and quantification process took less than 30 minutes on my computer, which is much faster than other tools like STAR or Cell Ranger.
Explore the Results
Prepare variables and use fishpond R package to load the results.
1myPath<- '/media/alfonso/data/nextflow_PBMCs/output/quant/pbmc5k_d3_quant/af_quant/'
2
3custom_format <- list("counts" = c("U", "S", "A"),
4 "spliced" = c("S"),
5 "unspliced" = c("U"),
6 "ambiguous" = c("A"))
7sce <- fishpond::loadFry(myPath,
8 outputFormat = custom_format)
This creates a SingleCellExperiment object where the counts assay contains the total counts of unspliced, spliced and ambiguous UMIs and each of them is also stored in a separate assay. If you are not familiar with SingleCellExperiment objects, take a look to my post Single cell Bioconductor's choice: SCE.
Spliced and unspliced assays will be handy if you plan to do RNA velocity analysis later on. Check my previous post on RNA velocity for more details.
Let's explore the object.
1sce
class: SingleCellExperiment
dim: 38606 157273
metadata(0):
assays(4): counts spliced unspliced ambiguous
rownames(38606): ENSG00000278704 ENSG00000274847 ... ENSG00000290841 ENSG00000288057
rowData names(1): gene_ids
colnames(157273): ATCCCGCTCGTGCCCT TGAAACGGTTAACGCT ... ACCGCTAGTTCAAGGA CTACATTCATCCGTTA
colData names(1): barcodes
reducedDimNames(0):
mainExpName: NULL
altExpNames(0):
the object has 38606 genes and 157273 cells, this means the droplets were not filtered by simpleaf. We will do it using the DropletUtils package.
1library(DropletUtils)
2bcrank <- barcodeRanks(counts(sce))
3
4# Only unique points for speed.
5uniq <- !duplicated(bcrank$rank)
6
7plot(bcrank$rank[uniq], bcrank$total[uniq], log="xy",
8 xlab="Rank", ylab="Total UMI count", cex.lab=1.2)
9abline(h=metadata(bcrank)$inflection, col="darkgreen", lty=2)
10abline(h=metadata(bcrank)$knee, col="dodgerblue", lty=2)
11legend("bottomleft", legend=c("Inflection", "Knee"),
12 col=c("darkgreen", "dodgerblue"), lty=2, cex=1.2)
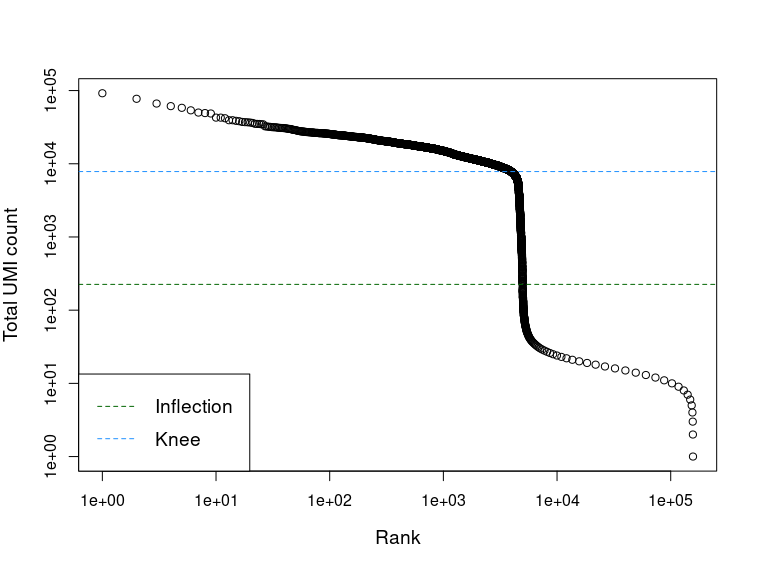
We can see that both the inflection and knee points are around 5000 droplets, the expected number of cells.
Conclusions
simpleafis a great tool for quantifying single cell RNA-seq data usingalevin-fry. It is fast and easy to use.- The use of a docker image simplifies the installation and avoids dependency issues. Making it even easier to use.
simpleafdoes not filter the droplets by default, keep this in mind if using it. This is easily solved using tools like theDropletUtilspackage.
Further reading
- Alevin-fry unlocks rapid, accurate, and memory-frugal quantification of single-cell RNA-seq data
- simpleaf documentation
- alevin-fry documentation
- "Single-cell best practices" book: Raw data processing
- “Orchestrating Single-Cell Analysis with Bioconductor” book: Droplet processing