Building Your First Nextflow Pipeline: PART 1 - One sample at a time
Nextflow is a workflow management system designed to simplify the process of writing and executing complex computational workflows. The main advantages of nextflow are:
- Reproducibility: ensures workflows can be run consistently across environments.
- Portability: supports execution on laptops, HPC clusters, cloud (AWS, GCP, Azure), and container platforms (Docker, Singularity).
- Scalability: handles anything from small local runs to massive parallel workloads.
- Modularity: pipelines are built from reusable processes that can be easily combined.
- Container support: seamless integration with Docker, Singularity, and Conda for reproducible software environments.
- Dataflow programming model: channels handle complex data parallelism naturally.
- Error handling & resume: automatic checkpointing allows workflows to resume from failure points.
In this post I will cover how to build your first pipeline. We will kind of follow this nextflow genomics tutorial.
Commands testing
The first step is testing the commands of the pipeline we wish to develop using the docker image. Since the idea is building a nextflow pipeline for alevin-fry so this step is already covered in my Pseudoalignment for single cell RNA-seq with alevin-fry post.
Single-stage workflow
We start creating a process called SIMPLEAF_INDEX describing the indexing step.
1/*
2 * Generate simpleaf index
3 */
4process SIMPLEAF_INDEX {
5
6 container 'quay.io/biocontainers/simpleaf:0.19.5--ha6fb395_0'
7
8 publishDir params.outdir, mode: 'symlink'
9
10 input:
11 path fasta
12 path gtf
13
14 output:
15 path "index_simpleaf"
16
17 script:
18 """
19 # export required var
20 export ALEVIN_FRY_HOME=.
21
22 # set maximum number of file descriptors for temp files
23 ulimit -n 2048
24
25 # prep simpleaf
26 simpleaf set-paths
27
28 simpleaf index \
29 --output simpleaf_index \
30 --fasta ${fasta} \
31 --gtf ${gtf} \
32 --threads 4 \
33 --work-dir ./workdir.noindex
34 """
35}
The process has 5 main blocks:
container: specifies the docker image to use for this processpublishDir: specifies where to save the output files
For this tutorial we will use
symlinkmode, which creates symbolic links to the output files in the specified directory. In general I use thecopymode so I do not need to keep the work folder (nexftlow internal folder) once the pipeline is done.
Check thepublishDirdocumentation for more details.
input: defines the input files or parameters required by the processoutput: defines the output files generated by the processscript: contains the actual commands to be executed
Next step is adding the input and output parameters. At the top of the file, under the Pipeline parameters section, we declare CLI parameters for development purposes.
1// Primary input
2params.fasta = "${projectDir}/reference/refdata-gex-GRCh38-2024-A/fasta/genome.fa"
3params.gtf = "${projectDir}/reference/refdata-gex-GRCh38-2024-A/genes/genes.gtf.gz"
4params.outdir = "results_genomics"
Finally, we add a workflow section at the end of the file to call the process. We just need to set up a channel for each input and then call the process.
1workflow {
2
3 // Create input channels
4 fasta_ch = Channel.fromPath(params.fasta)
5 gtf_ch = Channel.fromPath(params.gtf)
6
7 // Create index
8 SIMPLEAF_INDEX(
9 fasta_ch,
10 gtf_ch
11 )
12}
The last part is running the workflow we just created. I let you the task of building the whole post.1.nf script (the solution is on the bonus section). To run the workflow we use the following command:
1nexftlow run post.1.nf
Since our command runs in docker, we need to tell it to nextflow. This can be done in two ways:
- Adding
docker.enabled = trueto anextflow.configfile in the same directory as the script (I use this one)- Adding
-with-dockerto the command line
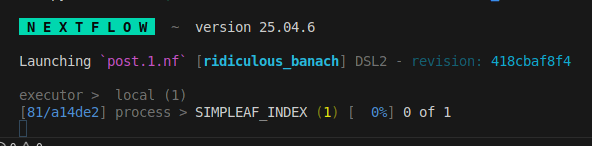
You should see something like this
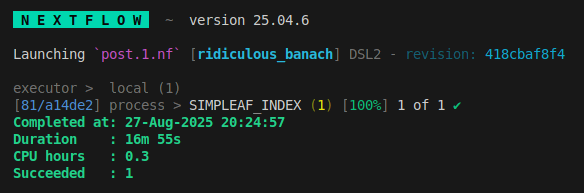
When it finishes you should see this
Add more processes
We start defining the next process, SIMPLEAF_QUANT, for quantification.
1/*
2 * Quantify Gene Expression
3 */
4process SIMPLEAF_QUANT {
5
6 container 'quay.io/biocontainers/simpleaf:0.19.5--ha6fb395_0'
7
8 publishDir params.outdir, mode: 'symlink'
9
10 input:
11 path index_path
12 path reads1_files
13 path reads2_files
14 val chemistry
15
16 output:
17 path "quant" , emit: quant_path
18
19 script:
20 def R1_FILES = reads1_files.collect().join(',')
21 def R2_FILES = reads2_files.collect().join(',')
22 """
23 # Download chemistry file
24 wget -O chemistries.json https://raw.githubusercontent.com/COMBINE-lab/simpleaf/dev/resources/chemistries.json || \
25 curl -o chemistries.json https://raw.githubusercontent.com/COMBINE-lab/simpleaf/dev/resources/chemistries.json
26
27 # export required var
28 export ALEVIN_FRY_HOME=.
29
30 # prep simpleaf
31 simpleaf set-paths
32
33 # run simpleaf quant
34 simpleaf quant \
35 --reads1 $R1_FILES \
36 --reads2 $R2_FILES \
37 --threads 4 \
38 --index ${index_path}/index \
39 --t2g-map ${index_path}/ref/t2g_3col.tsv \
40 --chemistry ${chemistry} \
41 --resolution cr-like \
42 --unfiltered-pl --anndata-out \
43 --output quant
44 """
45}
Now we add the needed parameters
1// Gene expression quantification
2params.sample_path = "${projectDir}/rawData/5k_Human_Donor3_PBMC_3p_gem-x_GEX_fastqs"
3params.chemistry = '10xv4-3p'
And we add the process call to the workflow section
1 // chemistry variable
2 chemistry = "${params.chemistry}"
3
4 // Get fastq files
5 reads1_files = Channel.fromPath("${params.sample_path}/*")
6 .filter { it.name.contains('_R1_') }
7 .collect()
8
9 reads2_files = Channel.fromPath("${params.sample_path}/*")
10 .filter { it.name.contains('_R2_') }
11 .collect()
12
13 // Quantify Gene Expression
14 quant = SIMPLEAF_QUANT(
15 SIMPLEAF_INDEX.out,
16 reads1_files,
17 reads2_files,
18 chemistry
19 )
To run the workflow, we use the same command as before but adding -resume. This way we do not repeat the steps that have already been done. Again, I challenge you to build the whole post.1.nf script (solution on the bonus section).
1nextflow run post.1.nf -resume
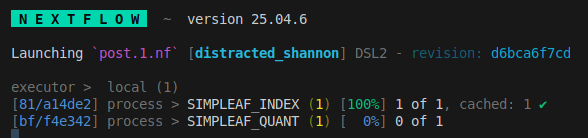
we see the index step is cached and has been skipped.
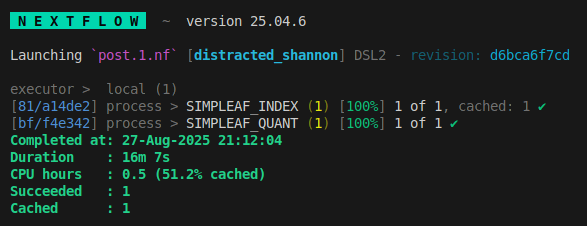
Now we can check the results, we use the -l flag to follow the symlinks
1tree -l results_simpleaf_index_test
1results_simpleaf_index_test
2├── index_simpleaf -> work/81/a14de2a6f5b378e66e9e21b444483a/index_simpleaf
3│ ├── index
4│ │ ├── gene_id_to_name.tsv
5│ │ ├── piscem_idx_cfish.json
6│ │ ├── piscem_idx.ctab
7│ │ ├── piscem_idx.ectab
8│ │ ├── piscem_idx.json
9│ │ ├── piscem_idx.refinfo
10│ │ ├── piscem_idx.sshash
11│ │ ├── simpleaf_index.json
12│ │ └── t2g_3col.tsv
13│ ├── index_info.json
14│ ├── ref
15│ │ ├── gene_id_to_name.tsv
16│ │ ├── roers_make-ref.json
17│ │ ├── roers_ref.fa
18│ │ └── t2g_3col.tsv
19│ └── simpleaf_index_log.json
20└── quant -> work/bf/f4e3421c875998196040a67c2fead5/quant
21 ├── af_map
22 │ ├── map_info.json
23 │ ├── map.rad
24 │ └── unmapped_bc_count.bin
25 ├── af_quant
26 │ ├── alevin
27 │ │ ├── quants.h5ad
28 │ │ ├── quants_mat_cols.txt
29 │ │ ├── quants_mat.mtx
30 │ │ └── quants_mat_rows.txt
31 │ ├── collate.json
32 │ ├── featureDump.txt
33 │ ├── gene_id_to_name.tsv
34 │ ├── generate_permit_list.json
35 │ ├── map.collated.rad
36 │ ├── permit_freq.bin
37 │ ├── permit_map.bin
38 │ ├── quant.json
39 │ ├── simpleaf_map_info.json
40 │ └── unmapped_bc_count_collated.bin
41 └── simpleaf_quant_log.json
This is it for today. We have develop a single-script nextflow pipeline that indexes the genome and processes a single hard-coded sample. In the next posts of this series we will see how to process multiple samples and how to split the code into modules to take full advantage of nextflow capabilities.
Bonus
View command
During development and testing the command view is useful to check channels and variables. Try to include the following examples in the workflow section
1SIMPLEAF_INDEX.out.view()
2
3reads1_files.view { "R1 files: ${it}" }
4reads2_files.view { "R2 files: ${it}" }
Indexing Nextflow Script
This is the complete nextflow script we have used to build the index
1// Primary input
2params.fasta = "${projectDir}/reference/refdata-gex-GRCh38-2024-A/fasta/genome.fa"
3params.gtf = "${projectDir}/reference/refdata-gex-GRCh38-2024-A/genes/genes.gtf.gz"
4params.outdir = "results_simpleaf_index_test"
5
6/*
7 * Generate simpleaf index
8 */
9process SIMPLEAF_INDEX {
10
11 container 'quay.io/biocontainers/simpleaf:0.19.5--ha6fb395_0'
12
13 publishDir params.outdir, mode: 'symlink'
14
15 input:
16 path fasta
17 path gtf
18
19 output:
20 path "index_simpleaf"
21
22 script:
23 """
24 # export required var
25 export ALEVIN_FRY_HOME=.
26
27 # set maximum number of file descriptors for temp files
28 ulimit -n 2048
29
30 # prep simpleaf
31 simpleaf set-paths
32
33 simpleaf index \
34 --output index_simpleaf \
35 --fasta ${fasta} \
36 --gtf ${gtf} \
37 --threads 4 \
38 --work-dir ./workdir.noindex
39 """
40}
41
42
43workflow {
44
45 // Create input channels
46 fasta_ch = Channel.fromPath(params.fasta)
47 gtf_ch = Channel.fromPath(params.gtf)
48
49 // Create index
50 SIMPLEAF_INDEX(
51 fasta_ch,
52 gtf_ch
53 )
54}
Full Nextflow Script
This is the final nextflow script of this post
1// Primary input
2params.fasta = "${projectDir}/reference/refdata-gex-GRCh38-2024-A/fasta/genome.fa"
3params.gtf = "${projectDir}/reference/refdata-gex-GRCh38-2024-A/genes/genes.gtf.gz"
4params.outdir = "results_simpleaf_index_test"
5
6// Gene expression quantification
7params.sample_path = "${projectDir}/rawData/5k_Human_Donor3_PBMC_3p_gem-x_GEX_fastqs"
8params.chemistry = '10xv4-3p'
9
10/*
11 * Generate simpleaf index
12 */
13process SIMPLEAF_INDEX {
14
15 container 'quay.io/biocontainers/simpleaf:0.19.5--ha6fb395_0'
16
17 publishDir params.outdir, mode: 'symlink'
18
19 input:
20 path fasta
21 path gtf
22
23 output:
24 path "index_simpleaf"
25
26 script:
27 """
28 # export required var
29 export ALEVIN_FRY_HOME=.
30
31 # set maximum number of file descriptors for temp files
32 ulimit -n 2048
33
34 # prep simpleaf
35 simpleaf set-paths
36
37 simpleaf index \
38 --output index_simpleaf \
39 --fasta ${fasta} \
40 --gtf ${gtf} \
41 --threads 4 \
42 --work-dir ./workdir.noindex
43 """
44}
45
46/*
47 * Quantify Gene Expression
48 */
49process SIMPLEAF_QUANT {
50
51 container 'quay.io/biocontainers/simpleaf:0.19.5--ha6fb395_0'
52
53 publishDir params.outdir, mode: 'symlink'
54
55 input:
56 path index_path
57 path reads1_files
58 path reads2_files
59 val chemistry
60
61 output:
62 path "quant" , emit: quant_path
63
64 script:
65 def R1_FILES = reads1_files.collect().join(',')
66 def R2_FILES = reads2_files.collect().join(',')
67 """
68 # Download chemistry file
69 wget -O chemistries.json https://raw.githubusercontent.com/COMBINE-lab/simpleaf/dev/resources/chemistries.json || \
70 curl -o chemistries.json https://raw.githubusercontent.com/COMBINE-lab/simpleaf/dev/resources/chemistries.json
71
72 # export required var
73 export ALEVIN_FRY_HOME=.
74
75 # prep simpleaf
76 simpleaf set-paths
77
78 # run simpleaf quant
79 simpleaf quant \
80 --reads1 $R1_FILES \
81 --reads2 $R2_FILES \
82 --threads 4 \
83 --index ${index_path}/index \
84 --t2g-map ${index_path}/ref/t2g_3col.tsv \
85 --chemistry ${chemistry} \
86 --resolution cr-like \
87 --unfiltered-pl --anndata-out \
88 --output quant
89 """
90}
91
92
93workflow {
94
95 // Create input channels
96 fasta_ch = Channel.fromPath(params.fasta)
97 gtf_ch = Channel.fromPath(params.gtf)
98
99 // chemistry variable
100 chemistry = "${params.chemistry}"
101
102 // Create index
103 SIMPLEAF_INDEX(
104 fasta_ch,
105 gtf_ch
106 )
107
108 // Get fastq files
109 reads1_files = Channel.fromPath("${params.sample_path}/*")
110 .filter { it.name.contains('_R1_') }
111 .collect()
112
113 reads2_files = Channel.fromPath("${params.sample_path}/*")
114 .filter { it.name.contains('_R2_') }
115 .collect()
116
117 // Quantify Gene Expression
118 quant = SIMPLEAF_QUANT(
119 SIMPLEAF_INDEX.out,
120 reads1_files,
121 reads2_files,
122 chemistry
123 )
124}