Alignment or Pseudoalignment: Alevin-fry vs Cell Ranger comparison
Last week post explained how to use simpleaf to run alevin-fry on a single cell RNA-seq dataset. This post will compare the results of alevin-fry with those of cellranger, the official 10x Genomics pipeline for single cell RNA-seq data analysis.
Let's start by checking the knee plot from the Cell Ranger html report.
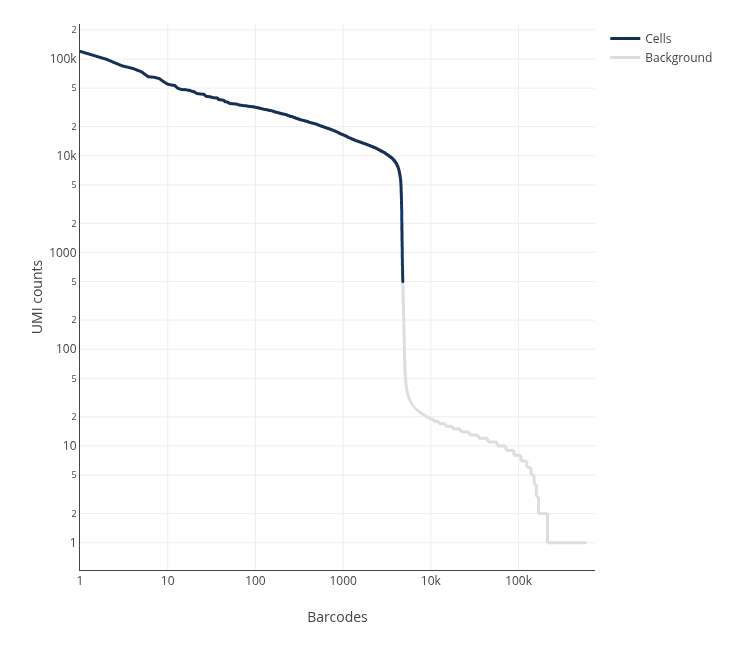
Now we can download Cell Ranger results from the 5k Human PBMCs (Donor 3) dataset website, this way we do not need to run Cell Ranger ourselves. We will use the filtered matrix, which is the result of selecting the cells labeled in blue in the knee plot.
1wget https://cf.10xgenomics.com/samples/cell-exp/9.0.0/5k_Human_Donor3_PBMC_3p_gem-x_5k_Human_Donor3_PBMC_3p_gem-x/5k_Human_Donor3_PBMC_3p_gem-x_5k_Human_Donor3_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.tar.gz
2tar -xvzf 5k_Human_Donor3_PBMC_3p_gem-x_5k_Human_Donor3_PBMC_3p_gem-x_count_sample_filtered_feature_bc_matrix.tar.gz
Next step is to read the files and create a SingleCellExperiment (SCE) object. If you are not familiar with SCE objects, take a look to my post Single cell Bioconductor's choice: SCE.
First we set variables
1matrix_dir_filtered <- 'sample_filtered_feature_bc_matrix'
2features.path <- paste0(matrix_dir_filtered, "/features.tsv.gz")
3barcodes.path <- paste0(matrix_dir_filtered, "/barcodes.tsv.gz")
4matrix.path <- paste0(matrix_dir_filtered, "/matrix.mtx.gz")
and then read the files to create the SCE object
1library( Matrix )
2library( SingleCellExperiment )
3mat <- readMM(file = matrix.path)
4feature.names = read.delim(features.path,
5 header = FALSE,
6 stringsAsFactors = FALSE)
7barcode.names = read.delim(barcodes.path,
8 header = FALSE,
9 stringsAsFactors = FALSE)
10colnames(mat) = barcode.names$V1
11rownames(mat) = feature.names$V1
12mat <- mat[ rowSums(mat) != 0, ]
13sce.cellranger <- SingleCellExperiment( assays = list( counts = mat ) )
check object
1sce.cellranger
1## class: SingleCellExperiment
2## dim: 27439 4782
3## metadata(0):
4## assays(1): counts
5## rownames(27439): ENSG00000238009 ENSG00000239945 ... ENSG00000275063
6## ENSG00000278817
7## rowData names(0):
8## colnames(4782): AAACCAAAGCGCCTTC-1 AAACCAAAGTTAATGC-1 ...
9## TGTGTTGAGCTTAGTG-1 TGTGTTGAGGAATGAC-1
10## colData names(0):
11## reducedDimNames(0):
12## mainExpName: NULL
13## altExpNames(0):
Now we can load the alevin-fry resulst using the fishpond R package as in my last post.
1myPath<- '/media/alfonso/data/nextflow_PBMCs/output/quant/pbmc5k_d3_quant/af_quant/'
2
3custom_format <- list("counts" = c("U", "S", "A"),
4 "spliced" = c("S"),
5 "unspliced" = c("U"),
6 "ambiguous" = c("A"))
7sce.alevin <- fishpond::loadFry(myPath,
8 outputFormat = custom_format)
1## locating quant file
1## Reading meta data
1## USA mode: TRUE
1## Processing 38606 genes and 157273 barcodes
1## Using user-defined output assays
1## Building the 'counts' assay, which contains U S A
1## Building the 'spliced' assay, which contains S
1## Building the 'unspliced' assay, which contains U
1## Building the 'ambiguous' assay, which contains A
1## Constructing output SingleCellExperiment object
1## Done
as we know, this object is not filtered. Let's filter it using the DropletUtils package.
1library(DropletUtils)
2bcrank <- barcodeRanks(counts(sce.alevin))
let's explore the results with a kneeplot
1uniq <- !duplicated(bcrank$rank)
2plot(bcrank$rank[uniq], bcrank$total[uniq], log="xy",
3 xlab="Rank", ylab="Total UMI count", cex.lab=1.2)
4abline(h=metadata(bcrank)$inflection, col="darkgreen", lty=2)
5abline(h=metadata(bcrank)$knee, col="dodgerblue", lty=2)
6legend("bottomleft", legend=c("Inflection", "Knee"),
7 col=c("darkgreen", "dodgerblue"), lty=2, cex=1.2)
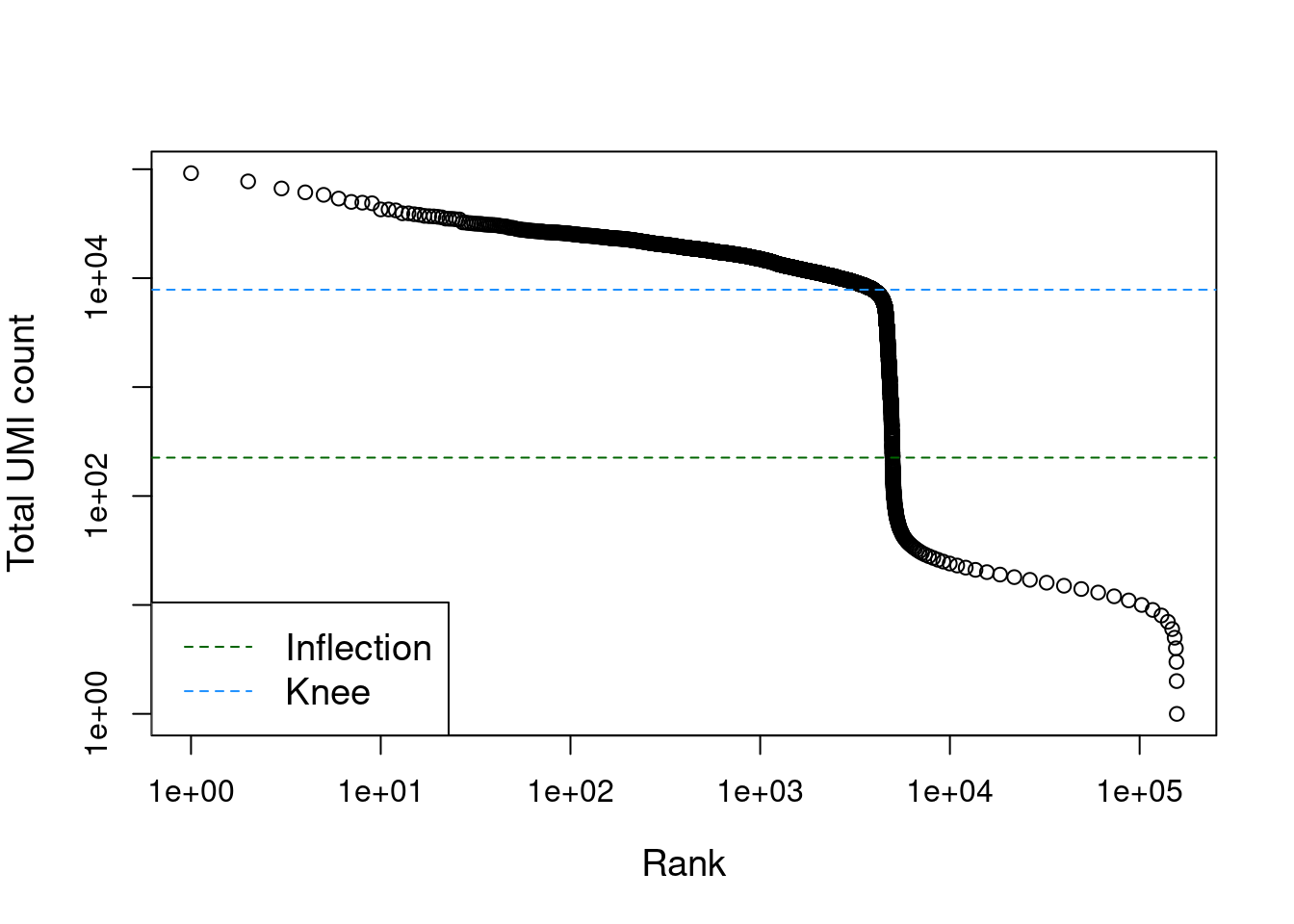
Now we can filter the alevin-fry object using the inflection point calculated by barcodeRanks function, we use the inflection point as it seems closer to the Cell Ranger's knee plot cell selection.
1sce.alevin <- sce.alevin[, bcrank$total >= metadata(bcrank)$inflection]
2sce.alevin <- sce.alevin[ rowSums(counts(sce.alevin)) > 0, ]
3sce.alevin
1## class: SingleCellExperiment
2## dim: 27249 4986
3## metadata(0):
4## assays(4): counts spliced unspliced ambiguous
5## rownames(27249): ENSG00000274847 ENSG00000278384 ... ENSG00000286130
6## ENSG00000290841
7## rowData names(1): gene_ids
8## colnames(4986): ATCCCGCTCGTGCCCT TGAAACGGTTAACGCT ... TCGCTCTGTTGCACCA
9## TGAGTAGGTGGCAGTG
10## colData names(1): barcodes
11## reducedDimNames(0):
12## mainExpName: NULL
13## altExpNames(0):
From the dimensions of both SCE objects, we can see that both objects have close to 5000 cells. However, the Cell Ranger object has around 4700 while the alevin-fry around 4900.
We use a Venn diagram to compare the cells in both objects
1library(VennDiagram)
2library(wesanderson)
3
4cells.list <- list( cellranger = gsub( '-1', '', colnames(sce.cellranger) ),
5 alevinfry = colnames(sce.alevin)
6 )
7
8venn.diagram( cells.list,
9 "Venn.diagram.tiff",
10 fill=wes_palette("FantasticFox1")[1:length(cells.list)],
11 alpha=rep( 0.25, length(cells.list) ),
12 cex = 2.5, cat.fontface=4,
13 category.names = names(cells.list),
14 col=wes_palette("FantasticFox1")[1:length(cells.list)]
15 )
16
17cmd <- 'convert Venn.diagram.tiff Venn.diagram.png && rm Venn.diagram.tiff '
18system( cmd )
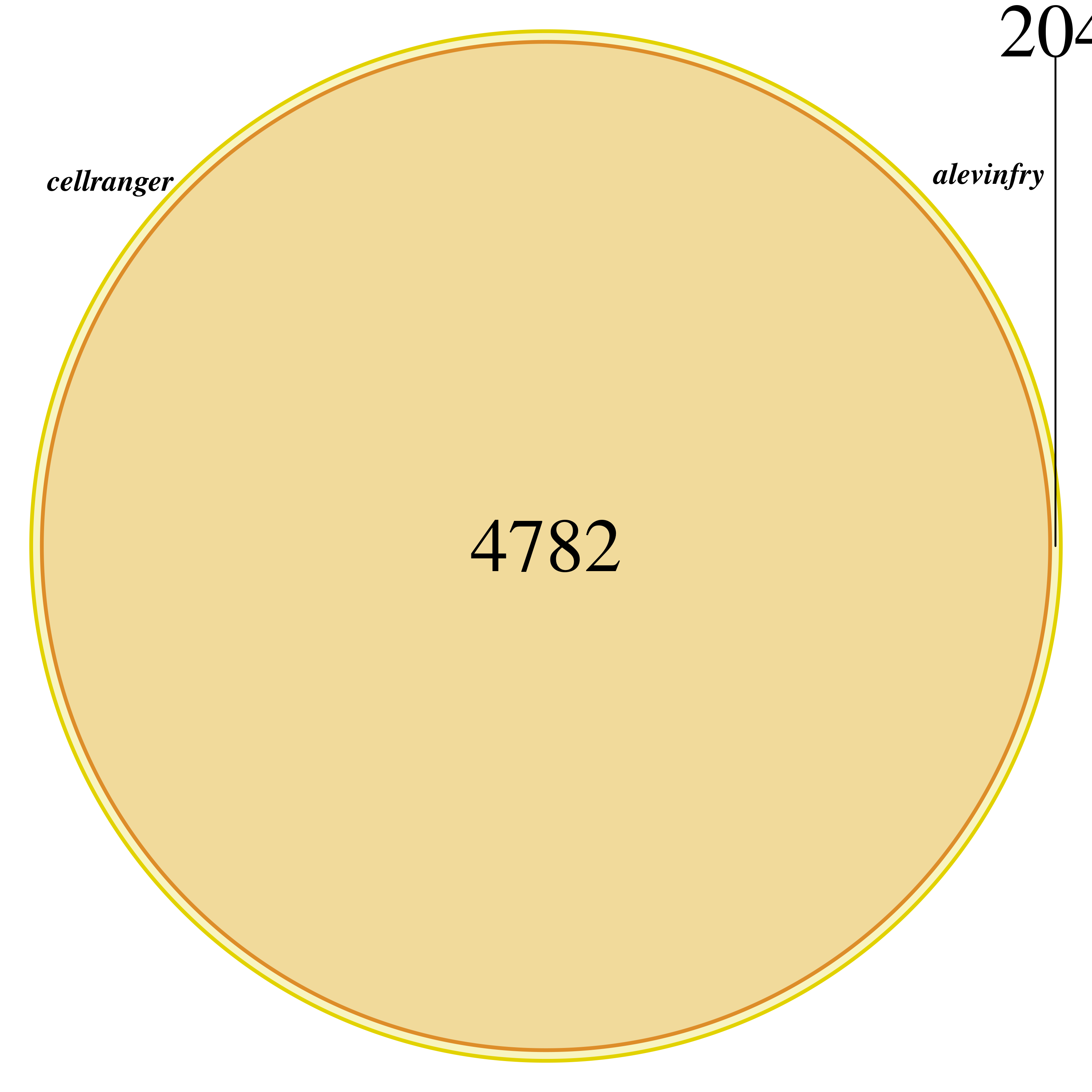
We see that all cells detected by Cell Ranger are also detected by alevin-fry, with alevin-fry detecting a few more.
This is most likely due to the barcodeRanks function's inflection point definition. In any case, this is not highly relevant as the first step recommended in any single cell workflow is filtering low quality cells using number of detected genes, number of UMIs, and percentage of mitochondrial genes.
Now we check the detected genes by each method.
1cells.list <- list( cellranger = rownames(sce.cellranger),
2 alevinfry = rownames(sce.alevin)
3 )
4
5venn.diagram( cells.list,
6 "Venn.rownames.tiff",
7 fill=wes_palette("FantasticFox1")[1:length(cells.list)],
8 alpha=rep( 0.25, length(cells.list) ),
9 cex = 2.5, cat.fontface=4,
10 category.names = names(cells.list),
11 col=wes_palette("FantasticFox1")[1:length(cells.list)]
12 )
13
14cmd <- 'convert Venn.rownames.tiff Venn.rownames.png && rm Venn.rownames.tiff '
15system( cmd )
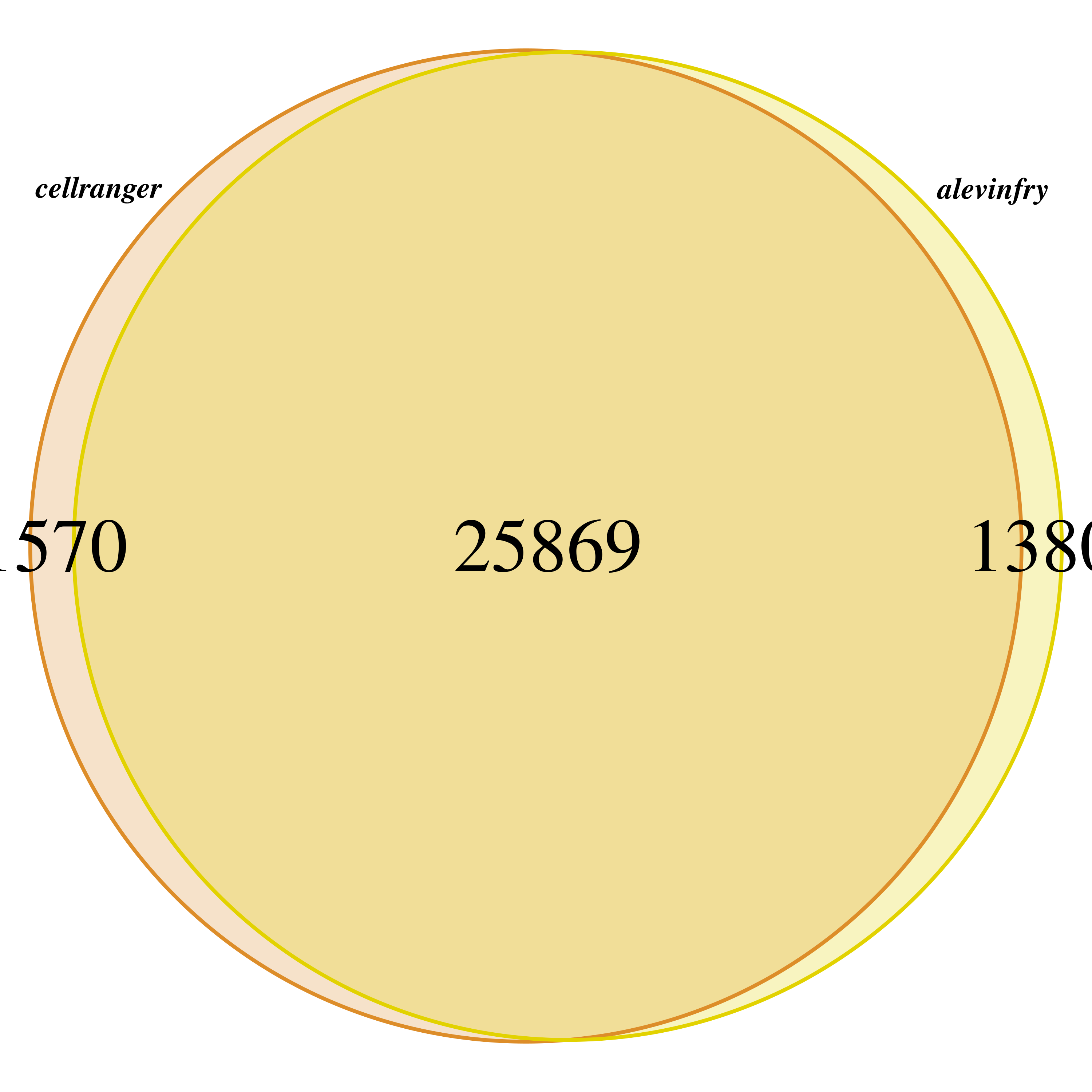
Again, very similar resuts, with over 25000 genes in common and only a little over 1000 genes detected specifically by one method.
Let's compare the expression in both methods with a scatter plot. In this plot each dot is the gene expression of a gene in a cell.
1library(ggplot2)
2library(dplyr)
1##
2## Attaching package: 'dplyr'
1## The following object is masked from 'package:Biobase':
2##
3## combine
1## The following objects are masked from 'package:GenomicRanges':
2##
3## intersect, setdiff, union
1## The following object is masked from 'package:GenomeInfoDb':
2##
3## intersect
1## The following objects are masked from 'package:IRanges':
2##
3## collapse, desc, intersect, setdiff, slice, union
1## The following objects are masked from 'package:S4Vectors':
2##
3## first, intersect, rename, setdiff, setequal, union
1## The following objects are masked from 'package:BiocGenerics':
2##
3## combine, intersect, setdiff, union
1## The following object is masked from 'package:matrixStats':
2##
3## count
1## The following objects are masked from 'package:stats':
2##
3## filter, lag
1## The following objects are masked from 'package:base':
2##
3## intersect, setdiff, setequal, union
1library(tidyr)
1##
2## Attaching package: 'tidyr'
1## The following object is masked from 'package:S4Vectors':
2##
3## expand
1## The following objects are masked from 'package:Matrix':
2##
3## expand, pack, unpack
1cr.counts.df <- counts(sce.cellranger) %>%
2 as.matrix() %>%
3 as.data.frame() %>%
4 tibble::rownames_to_column(var = "gene") %>%
5 pivot_longer(-gene, names_to = "cell", values_to = "cellranger") %>%
6 mutate(cell = gsub( '-1', '', cell ))
7
8alevin.counts.df <- counts(sce.alevin) %>%
9 as.matrix() %>%
10 as.data.frame() %>%
11 tibble::rownames_to_column(var = "gene") %>%
12 pivot_longer(-gene, names_to = "cell", values_to = "alevin")
1## Warning in asMethod(object): sparse->dense coercion: allocating vector of size
2## 1.0 GiB
1plot.df <- cr.counts.df %>%
2 full_join(alevin.counts.df, by = c("gene", "cell")) %>%
3 replace_na(list(alevin = 0, cellranger = 0)) %>%
4 filter( !( cellranger == 0 & alevin == 0 ) )
5
6ggplot(plot.df, aes(x = cellranger, y = alevin)) +
7 geom_abline(slope = 1, intercept = 0, color = "navy") +
8 geom_point(alpha = 0.1) +
9 # scale_x_log10() +
10 # scale_y_log10() +
11 labs(x = "Cell Ranger Counts", y = "Alevin-Fry Counts") +
12 theme_bw() +
13 theme(legend.position = "none")
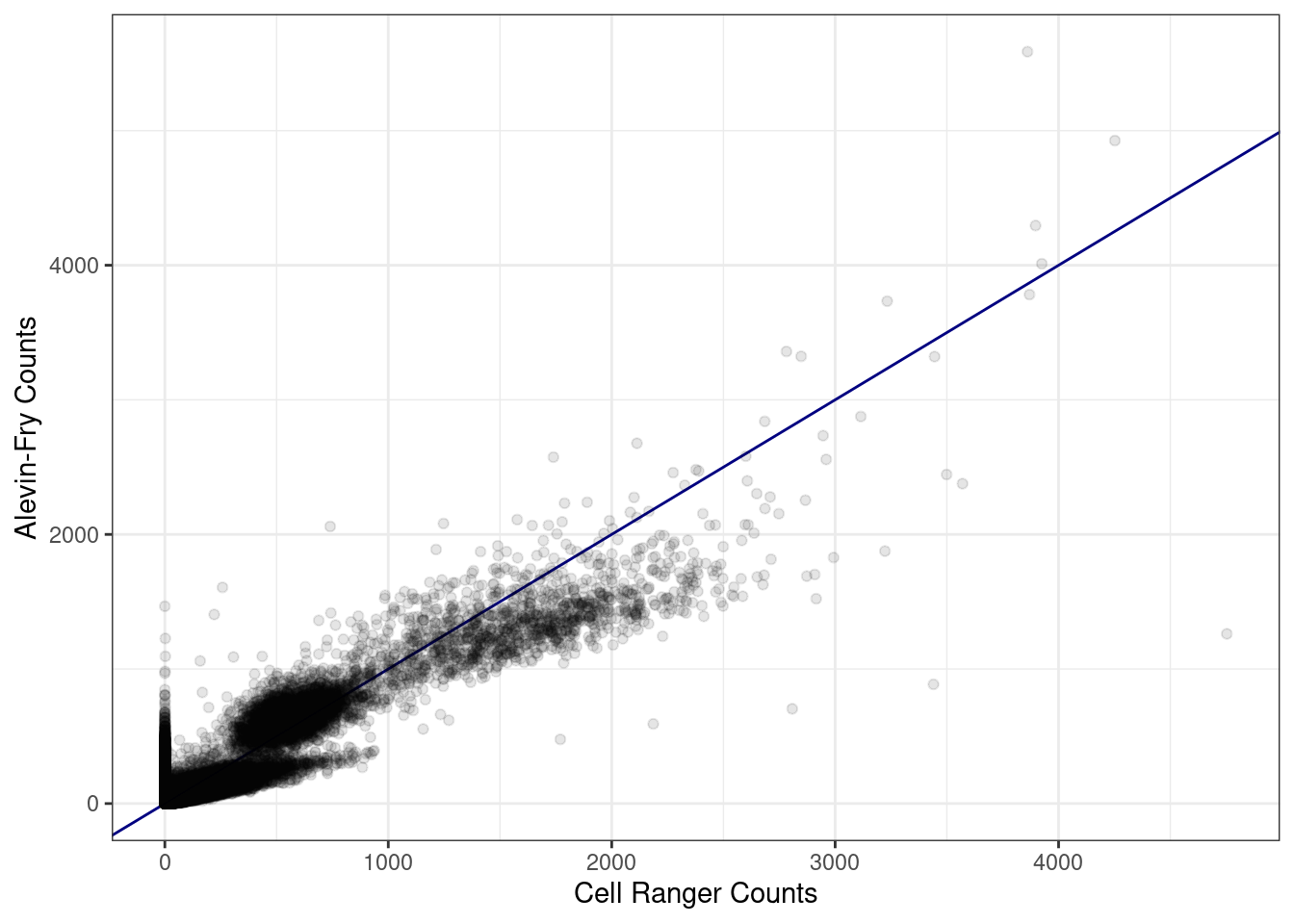
In general, most genes are detected at 'comparable' levels by both methods. We already knew from the Venn diagram analysis that a little over 1000 genes were only detected by one of the methods. The scatter plots shows that the genes only detected by alevin-fry have a significant expression, that's why we see that column of cells at x = 0.
We start by quantifying genes that not detected by cellranger but detected by alevin-fry.
1plot.df %>%
2 filter( cellranger == 0 & alevin > 0 ) %>%
3 select(gene) %>%
4 distinct() %>%
5 nrow()
1## [1] 26694
most genes are detected in at least one cell by alevin-fry but not cellranger. This is quite expected due to the known dropout effect in single cell RNAseq.
Let's try to quantify the number of cells per gene.
1summary <- plot.df %>%
2 filter( cellranger == 0 & alevin > 0 ) %>%
3 group_by(gene) %>%
4 summarise( n_cells = n(), max = max(alevin), avg = mean(alevin), median = median(alevin), min = min(alevin) )
Let's take a look to the genes detected in the higher number of cells.
1summary %>%
2 arrange(desc(n_cells)) %>%
3 head(20)
1## # A tibble: 20 × 6
2## gene n_cells max avg median min
3## <chr> <int> <dbl> <dbl> <dbl> <dbl>
4## 1 ENSG00000101333 4931 90 12.3 11 1
5## 2 ENSG00000247134 4769 51 8.55 8 1
6## 3 ENSG00000228716 4718 1466 220. 203 1
7## 4 ENSG00000178568 4410 21 3.10 3 1
8## 5 ENSG00000280441 4366 427 68.0 60 1
9## 6 ENSG00000163590 4005 123 18.9 18 1
10## 7 ENSG00000211592 3887 12 2.71 2 1
11## 8 ENSG00000125538 3684 33 3.56 3 1
12## 9 ENSG00000163220 3588 63 6.79 6 1
13## 10 ENSG00000038427 3577 38 4.39 4 1
14## 11 ENSG00000169429 3559 20 3.26 3 1
15## 12 ENSG00000236383 3513 31 2.75 2 1
16## 13 ENSG00000090382 3502 148 16.9 16 1
17## 14 ENSG00000123689 3479 30 3.93 3 1
18## 15 ENSG00000101439 3478 41 5.10 5 1
19## 16 ENSG00000153823 3390 24 3.11 3 1
20## 17 ENSG00000143546 3369 17 2.98 3 1
21## 18 ENSG00000085265 3353 24 3.30 3 1
22## 19 ENSG00000211899 3342 10 2.04 2 1
23## 20 ENSG00000124882 3310 22 2.75 2 1
Now let's focus on those genes with highest averagte expression.
1summary %>%
2 arrange(desc(avg)) %>%
3 head(20)
1## # A tibble: 20 × 6
2## gene n_cells max avg median min
3## <chr> <int> <dbl> <dbl> <dbl> <dbl>
4## 1 ENSG00000228716 4718 1466 220. 203 1
5## 2 ENSG00000280441 4366 427 68.0 60 1
6## 3 ENSG00000251562 216 619 57.3 45 8
7## 4 ENSG00000163590 4005 123 18.9 18 1
8## 5 ENSG00000090382 3502 148 16.9 16 1
9## 6 ENSG00000167996 363 83 16.1 10 1
10## 7 ENSG00000133112 387 198 15.7 10 1
11## 8 ENSG00000137818 383 144 15.0 8 1
12## 9 ENSG00000156508 316 162 14.8 9 1
13## 10 ENSG00000198804 205 53 14.6 13 1
14## 11 ENSG00000167526 362 104 14.2 8 1
15## 12 ENSG00000142937 401 111 13.9 8 1
16## 13 ENSG00000147403 396 126 13.7 8 1
17## 14 ENSG00000112306 425 87 13.2 7 1
18## 15 ENSG00000101333 4931 90 12.3 11 1
19## 16 ENSG00000142541 399 109 11.7 7 1
20## 17 ENSG00000164587 456 96 11.6 7 1
21## 18 ENSG00000019582 1173 32 11.6 12 1
22## 19 ENSG00000145425 387 104 11.2 6 1
23## 20 ENSG00000169554 2395 41 11.2 11 1
Checking a few of the genes detected in most cells at significant levels, we find:
- ENSG00000228716, a dihydrofolate reductase whose deficiency has been linked to megaloblastic anemia. Several transcript variants encoding different isoforms have been found for this gene.
- ENSG00000280441, a lncRNA with little information available.
- ENSG00000163590, a magnesium or manganese-requiring phosphatase that is involved in several signaling pathways
it would be interesting to check if gene detected in lower number of cells are associated with specific cell types or clusters.
Conclusions
- Both methods produce similar results.
- It would be interesting to evaluate the expression of 'method specific' genes, specially in the context of cell type or cluster specific expression.
- It would also be interesting to check if these minor differences have an effect on downstream analyses.
Further reading
- 10X Genomics: Cell Ranger
- "Single-cell best practices" book: Quality Control
- “Orchestrating Single-Cell Analysis with Bioconductor” book: Chapter 1 Quality Control
- Seurat - Guided Clustering Tutorial: QC and selecting cells for further analy