Building Your First Nextflow Pipeline: PART 4 - Polish your pipeline
Nextflow is a workflow management system designed to simplify the process of writing and executing complex computational workflows. The main advantages of nextflow are:
- Reproducibility: ensures workflows can be run consistently across environments.
- Portability: supports execution on laptops, HPC clusters, cloud (AWS, GCP, Azure), and container platforms (Docker, Singularity).
- Scalability: handles anything from small local runs to massive parallel workloads.
- Modularity: pipelines are built from reusable processes that can be easily combined.
- Container support: seamless integration with Docker, Singularity, and Conda for reproducible software environments.
- Dataflow programming model: channels handle complex data parallelism naturally.
- Error handling & resume: automatic checkpointing allows workflows to resume from failure points.
Following the previous posts of this series (workflow that only processes one sample, multiple samples and Modularization & Configurable settings) we constructed a fully functional pipeline. Today, we will polish it to make it more professional and shareable.
Workflow outputs
Our current pipeline uses publishDir to move the results of each process to the desired output directory. For example, in the INDEX process:
1publishDir params.outdir, mode: 'symlink'
Last versions of nextflow (>=24.04.0) introduced a new way to define workflow outputs using the output block in the main workflow script. This approach is more flexible and powerful than publishDir, allowing you to specify exactly which files or channels should be treated as outputs of the workflow. See here how to migrate from publishDir to workflow outputs.
We start creating a new main block at the beginning of the workflow:
1workflow {
2 main:
3 // define input files & variables
4 ...
add a publish block in the workflow and a output section at the end of the script. Note that we an specify the mode as we did for the publishDir directive.
1 ...
2 publish:
3 simpleaf_index = index.index_path
4 quantification_results = quant.quant_path
5}
6
7output {
8 simpleaf_index {
9 path "${params.outdir}/'index"
10 mode 'symlink'
11 }
12
13 quantification_results {
14 path "${params.outdir}/'quant'"
15 mode 'symlink'
16 }
17}
This feature requires the nextflow.preview.output feature flag to be enabled. I prefer to add it before the ouput block for clarity:
1 ...
2 }
3
4// Workflow outputs
5nextflow.preview.output = true
6
7output {
8 ...
All files in the published channels are copied into the output directory, which is results by default. The output directory can be set using the outputDir config setting or the -output-dir command-line option.
to keep the outdir paramater, we can add the following line to the nextflow.config file:
1// Set outputDir based on parameter (with default)
2outputDir = params.outdir
Last thing is removing all the publishDir directives from the processes.
Now we can test the workflow, with -resume it should not need to do anything.
1nextflow run post.5.nf -resume
According to nextflow developers, this approach is intended to replace the publishDir directive so it would be wise to start using it now. Just stay tuned to the official documentation for updates as this is a new feature and may evolve as said in the image.
help
IF you want to share your pipeline or just for documentation (your future you will thank you), it is a good practice to add a help message to guide users on how to run it and what parameters are required.
Nextflow's official documentation doesn't provide a specific guide for adding help messages to pipelines. Using the params.help to show help messages is a community-established best practice, particularly popularized by nf-core.
Groovy is a programming language based on Java. Nextflow scripts are written in Groovy, so we will use groovy files as follows to implement the help functionality. Create the following file help.groovy within a help folder in the main folder of the pipeline
1def helpMessage(Map params) {
2 return """
3 ========================================
4 SimpleAF Single-Cell RNA-seq Pipeline
5 ========================================
6
7 Usage:
8 nextflow run main.nf [options]
9
10 Required Parameters:
11 --referenceFASTA Path to reference FASTA file
12 --referenceGTF Path to reference GTF file
13 --sample_info Path to sample information TSV file
14
15 Optional Parameters:
16 --outdir Output directory
17 --help Show this help message and exit
18
19 Examples:
20 nextflow run main.nf --help
21 nextflow run main.nf --sample_info samples.tsv --outdir my_results
22
23 Sample Info File Format:
24 The sample_info TSV file should contain the following columns:
25 - sample_id: Unique sample identifier
26 - chemistry: 10x chemistry (e.g., 10xv4-3p)
27 - sample_path: Path to directory containing FASTQ files
28 - reads1_pat: Pattern for R1 files (e.g., *R1*)
29 - reads2_pat: Pattern for R2 files (e.g., *R2*)
30
31 Output Structure:
32 Output_folder/
33 ├── index/
34 │ └── index_simpleaf/
35 └── quant/
36 ├── sample1_quant/
37 ├── sample2_quant/
38 """
39}
then add the following to the beginning of the main script:
1// Help setting
2params.help = false
3include { helpMessage } from './lib/help.groovy'
and this within the workflow, at the beginning:
1
2// Show help message if --help is provided
3if (params.help) {
4 println( helpMessage(params) )
5 exit 0
6}
now, before running your pipeline, you can check the help message with
1nextflow run post.5.nf --help
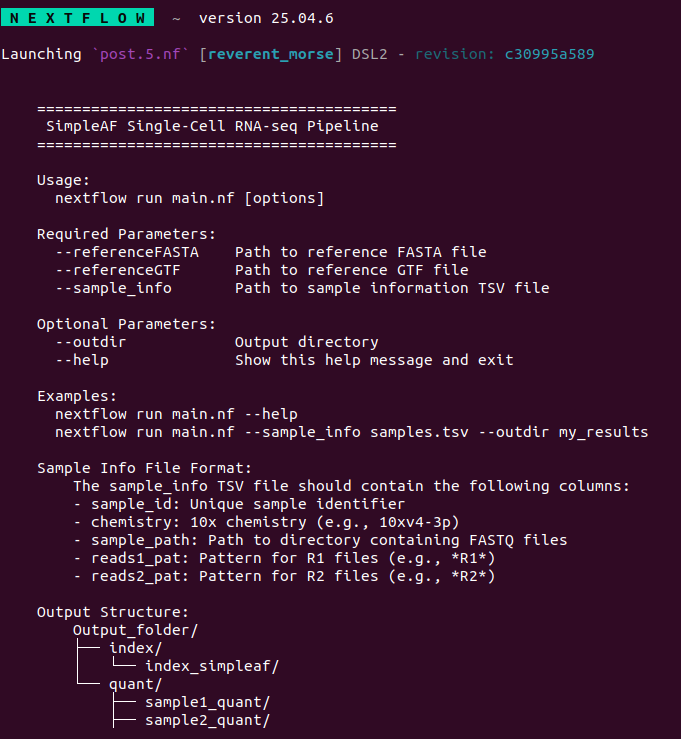
Parameter validation
The last part, maybe, to make your pipeline more professional and shareable is to validate the required parameters. Just add the following within the workflow, after the if (params.help) block:
1// Validate required parameters
2if (!params.referenceFASTA || !params.referenceGTF || !params.sample_info) {
3 error "Missing required parameters. Use --help for usage information."
4}
This is a very quick and simple validation, you can make it more complex to check each parameter individually and provide more specific error messages.
BONUS
Final pipeline
Folder structure
1.
2├── lib
3│ └── help.groovy
4├── modules
5│ └── simpleaf
6│ ├── index
7│ │ └── main.nf
8│ └── quant
9│ └── main.nf
10├── nextflow.config
11├── post.1.nf
12├── post.2.nf
13├── post.3.nf
14├── post.5.nf
15└── post.4.nf
post.5.nf
This would be the main.nf file of the final pipeline
1// Help setting
2params.help = false
3include { helpMessage } from './lib/help.groovy'
4
5// Include modules
6include { SIMPLEAF_INDEX as INDEX } from './modules/simpleaf/index/main.nf'
7include { QUANT as QUANT } from './modules/simpleaf/quant/main.nf'
8
9workflow {
10
11 // Show help message if --help is provided
12 if (params.help) {
13 println( helpMessage(params) )
14 exit 0
15 }
16
17 // Parameter validation
18 if (!params.outdir || !params.referenceFASTA || !params.referenceGTF || !params.sample_info) {
19 error "Missing required parameters. Use --help for usage information."
20 }
21
22 // define input files & variables
23 fasta = file("${params.referenceFASTA}") // reference fasta
24 gtf = file("${params.referenceGTF}") // annotation GTF
25
26 // Load sample information from TSV file
27 sample_info = Channel.fromPath(params.sample_info)
28 .splitCsv(header: true, sep: '\t')
29
30 // INDEX creation
31 index = INDEX(
32 fasta,
33 gtf
34 )
35
36 // prep for quantification
37 quant_input = sample_info
38 .map { x ->
39 def files_R1 = file("${x.sample_path}/*${x.reads1_pat}*", checkIfExists: true)
40 def files_R2 = file("${x.sample_path}/*${x.reads2_pat}*", checkIfExists: true)
41 tuple(x.sample_id, x.chemistry, files_R1, files_R2)
42 }
43
44 // Alevin-fry quantification
45 quant = QUANT(
46 index.index_path,
47 quant_input
48 )
49
50 publish:
51 simpleaf_index = index.index_path
52 quantification_results = quant.quant_path
53}
54
55// Workflow outputs
56nextflow.preview.output = true
57
58output {
59 simpleaf_index {
60 path "index"
61 mode 'symlink'
62 }
63
64 quantification_results {
65 path "quant"
66 mode 'symlink'
67 }
68}
Config file
1// setting parameters
2params {
3 referenceFASTA = 'reference/refdata-gex-GRCh38-2024-A/fasta/genome.fa'
4 referenceGTF = 'reference/refdata-gex-GRCh38-2024-A/genes/genes.gtf.gz'
5 sample_info = 'sample_info.tsv'
6 outdir = 'results_full'
7}
8
9// Enable Docker
10docker {
11 enabled = true
12}
13
14// Set outputDir based on parameter (with default)
15outputDir = params.outdir
Help file
1def helpMessage(Map params) {
2 return """
3 ========================================
4 SimpleAF Single-Cell RNA-seq Pipeline
5 ========================================
6
7 Usage:
8 nextflow run main.nf [options]
9
10 Required Parameters:
11 --referenceFASTA Path to reference FASTA file
12 --referenceGTF Path to reference GTF file
13 --sample_info Path to sample information TSV file
14
15 Optional Parameters:
16 --outdir Output directory
17 --help Show this help message and exit
18
19 Examples:
20 nextflow run main.nf --help
21 nextflow run main.nf --sample_info samples.tsv --outdir my_results
22
23 Sample Info File Format:
24 The sample_info TSV file should contain the following columns:
25 - sample_id: Unique sample identifier
26 - chemistry: 10x chemistry (e.g., 10xv4-3p)
27 - sample_path: Path to directory containing FASTQ files
28 - reads1_pat: Pattern for R1 files (e.g., _R1_)
29 - reads2_pat: Pattern for R2 files (e.g., _R2_)
30
31 Output Structure:
32 Output_folder/
33 ├── index/
34 │ └── index_simpleaf/
35 └── quant/
36 ├── sample1_quant/
37 ├── sample2_quant/
38 """
39}
Processes
INDEX process
1/*
2 * Generate simpleaf index
3 */
4process SIMPLEAF_INDEX {
5
6 container 'quay.io/biocontainers/simpleaf:0.19.5--ha6fb395_0'
7
8 // publishDir params.outdir, mode: 'symlink'
9
10 input:
11 path fasta
12 path gtf
13
14 output:
15 path "index_simpleaf", emit : index_path
16
17 script:
18 """
19 # export required var
20 export ALEVIN_FRY_HOME=.
21
22 # set maximum number of file descriptors for temp files
23 ulimit -n 2048
24
25 # prep simpleaf
26 simpleaf set-paths
27
28 simpleaf index \
29 --output index_simpleaf \
30 --fasta ${fasta} \
31 --gtf ${gtf} \
32 --threads 4 \
33 --work-dir ./workdir.noindex
34 """
35}
QUANT process
1/*
2 * Quantify Gene Expression
3 */
4process QUANT {
5
6 container 'quay.io/biocontainers/simpleaf:0.19.5--ha6fb395_0'
7
8 // publishDir "${params.outdir}/quant", mode: 'symlink'
9
10 input:
11 path index_path
12 tuple val(sample_id), val(chemistry), path(reads1_files), path(reads2_files)
13
14 output:
15 path "${sample_id}_quant" , emit: quant_path
16
17 script:
18 def R1_FILES = reads1_files.collect().join(',')
19 def R2_FILES = reads2_files.collect().join(',')
20 """
21 # Download chemistry file
22 wget -O chemistries.json https://raw.githubusercontent.com/COMBINE-lab/simpleaf/dev/resources/chemistries.json
23
24 # export required var
25 export ALEVIN_FRY_HOME=.
26
27 # prep simpleaf
28 simpleaf set-paths
29
30 # run simpleaf quant
31 simpleaf quant \
32 --reads1 $R1_FILES \
33 --reads2 $R2_FILES \
34 --threads 4 \
35 --index ${index_path}/index \
36 --t2g-map ${index_path}/ref/t2g_3col.tsv \
37 --chemistry ${chemistry} \
38 --resolution cr-like \
39 --unfiltered-pl --anndata-out \
40 --output ${sample_id}_quant
41 """
42}