From Prompt to Pipeline: Building a scRNA-seq Workflow with Claude Code and Nextflow
Building a complete scRNA-seq pipeline is a complex task that traditionally takes days or weeks of coding, debugging, and iterating. With Claude Code, you can focus on the biological questions like which integration method best preserves your cell populations, which clustering resolution captures meaningful heterogeneity, whether your DE results make biological sense while Claude handles the implementation. It's not about replacing bioinformatics expertise; it's about amplifying it. You still need to know what to ask for and why, but Claude Code takes care of the how.
For today's post, as a demo and practice, I decided to build on my previous post series on how to build NextFlow pipelines (workflow that only processes one sample, multiple samples, Modularization & Configurable settings, and Polish your pipeline) to build a NextFlow pipeline that performs the following steps:
- Quantification: simpleaf/alevin-fry for rapid quantification
- Per-sample QC: Scanpy-based quality control, filtering, and normalization
- Batch Integration: BBKNN for batch-corrected integration
This post assumes you already have Claude Code installed and configured. If you don't, check out my previous post on how to install and set up Claude Code for bioinformatics.
Project Setup
Before we start asking Claude to build anything, let's set up our project properly. Good project structure is half the battle in bioinformatics, and Claude Code works best when it understands your project layout from the start.
First, create your project directory and initialize Claude Code:
1mkdir claude_NF_scRNAseq
2cd claude_NF_scRNAseq
3claude
Once inside Claude Code, run the /init command to create the claude.md file:
1/init
This is where things get interesting. Remember from the previous post that the claude.md file gives Claude the domain-specific context it needs to work effectively. For single-cell analysis, this context is critical. Ask Claude to set it up with a prompt like this one:
1Please update the claude.md file for a single-cell RNA-seq analysis project with the following specifications:
2- We are working with 10X Genomics Chromium data (3' gene expression)
3- Reference genome: GRCh38 (human)
4- Primary analysis tools: Cell Ranger for alignment, Seurat (R) for downstream analysis
5- Python tools: Scanpy for alternative analyses and plotting
6- We follow the Satija lab best practices for QC thresholds
7- Directory structure: data/raw, data/processed, scripts/, results/figures, results/tables
8- Sample naming convention: {condition}_{replicate}_{lane} (e.g., treated_rep1_L001)
9- We prefer ggplot2 for publication-quality figures
10- All R scripts should use Seurat v5 syntax
11- All Python scripts should be compatible with Scanpy >= 1.10
Claude will generate a comprehensive claude.md file. Review it and refine as needed. You can always add more context or provide your desired context using the #add to memory command:
1#add to memory We have 4 samples: 2 control (ctrl_rep1, ctrl_rep2) and 2 treated (treated_rep1, treated_rep2), each sequenced on a single lane of a NovaSeq 6000
Creating the prompt
For this test I decided to push Claude Code to its limits and leave the claude.md empty while providing a very detailed initial prompt.
To generate a detailed and comprehensive prompt, I used another LLM (perplexity). I used the following prompt:
1hi perplexity, help write a detailed instruction for claude code. I want to build a nextflow pipeline that processes 10X datasets with alevin-fry using simpleaf, then reads the outputs on scanpy, performs stardard quality control on each sample independently and finally integrates them using BBKNN. What else do you need me to tell you to start?
Perplexity asked me several questions on multiple aspects from 'Dataset and inputs' and 'Reference and simpleaf/alevin‑fry settings' to 'Scanpy QC design', 'NextFlow' or 'Reproducibility and testing'. Following my answers it provided a detailed md file, you can find it on the pipeline repo on my github as prompt.qmd file.
It provides details in key aspects including the following
- Inputs
- Configurable parameters
- Environment and containers
- Directory structure
- basic QC metrics
- filter cells
- optional gene filtering
- doublet detection with scDblFinder (details depend on implementation)
- scale and PCA
- BBKNN neighbours
- UMAP and Leiden clustering
- basic plots
- Nextflow implementation details
- Examples on
config.filesandcode snipets.
Building the pipeline
With the prompt ready, I went to positron and launched claude in one of the terminals, started the plan mode (remember, Shift+Tab twice), and pasted the whole prompt.
Claude asked me a few questions (see them below with my answers):
- Which samplesheet format should we use for specifying FASTQ files?
- Which container should we use for the Scanpy/BBKNN processes?
- How should we handle doublet detection with scDblFinder?
- Do you have test data ready, or should I create example data structures?
After a couple of iterations to correct some details I accepted the plan and let Claude work on Auto-accept mode. It started to build everything and then testing it with some test data prepared by Claude.
However, Claude Code was not able to build the pipeline by itself. It could not fully define the correct commands for simpleaf, after a few iterations I decided to give Claude Code the links to my NextFlow post as example of a functional pipeline. I also had to manually tell Claude Code the proper Docker image for scanpy and BBKNN steps. Then runs failed because errors in the python code like using wrong packages or inaccurate code. Most python errors were fixed by Claude Code as follows
1try:
2 # Try with metric parameter (older scanpy versions)
3 sc.tl.umap(adata, min_dist=umap_min_dist, spread=umap_spread, metric=umap_metric)
4 logger.info(f"Computed UMAP with min_dist={umap_min_dist}, spread={umap_spread}, metric={umap_metric}")
5except TypeError as e:
6 # Fallback if metric parameter not supported (newer versions)
7 if 'metric' in str(e):
8 logger.warning(f"UMAP metric parameter not supported, computing without it: {e}")
9 sc.tl.umap(adata, min_dist=umap_min_dist, spread=umap_spread)
10 logger.info(f"Computed UMAP with min_dist={umap_min_dist}, spread={umap_spread}")
11 else:
12 raise
although it may be a good programming practice, in bioinformatics is not really needed to adapt the code to older versions. And many of the problems it found were related to using old code/versions.
Code repository
Claude was nice enough to produce a README.md so I initialized a git repo on the folder and ask Claude to commit all changes.
Then I pushed it to my github so you can all check and use it.
Running the pipeline
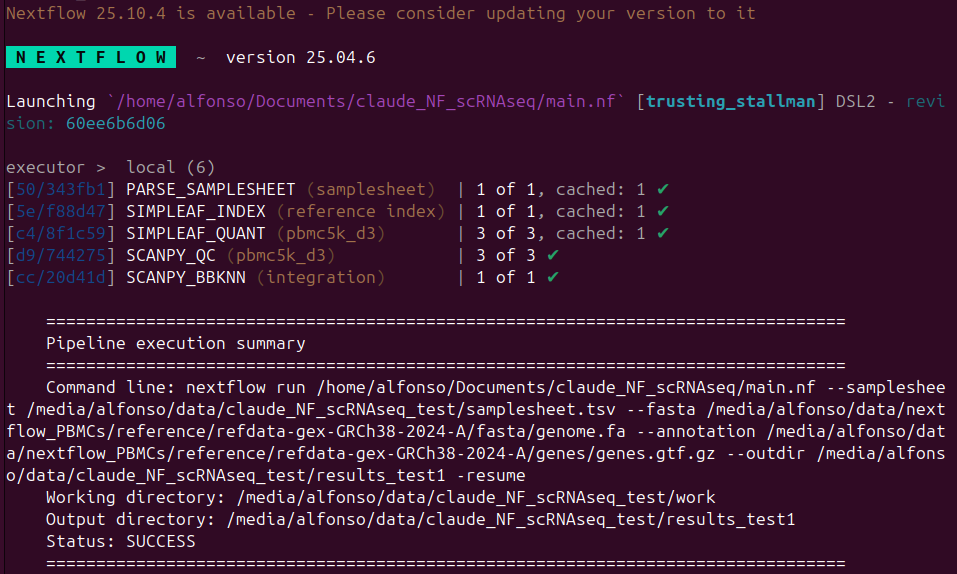
When Claude Code finally completed the development, the pipeline can be run as follows
1nextflow run .../claude_NF_scRNAseq/main.nf \
2 --samplesheet .../claude_NF_scRNAseq_test/samplesheet.tsv \
3 --fasta .../reference/refdata-gex-GRCh38-2024-A/fasta/genome.fa \
4 --annotation .../reference/refdata-gex-GRCh38-2024-A/genes/genes.gtf.gz \
5 --outdir .../claude_NF_scRNAseq_test/results_test1
where the --fasta and --annotation parameters point to the same files used in my previous post and the --samplesheet looks like this
1sample_id fastq_dir chemistry batch donor condition
2pbmc_1k .../pbmc_1k_v3_fastqs 10xv3 b1 d1 normal
3pbmc5k_d3 .../5k_Human_Donor3_PBMC_3p_gem-x_GEX_fastqs 10xv4-3p b3 d3 normal
4pbmc5k_d2 .../5k_Human_Donor2_PBMC_3p_gem-x_GEX_fastqs 10xv4-3p b2 d2 normal

The output folder has the following content
1results_test1/
2 ├── integration
3 │ └── bbknn
4 │ ├── merged_bbknn.h5ad
5 │ ├── umap_by_barcodes.png
6 │ ├── umap_by_batch.png
7 │ ├── umap_by_leiden.png
8 │ └── umap_by_sample_id.png
9 ├── parse
10 │ └── parsed_samples.tsv
11 ├── reports
12 │ ├── execution_timeline.html
13 │ └── execution_trace.txt
14 ├── scanpy
15 │ └── qc
16 │ ├── pbmc_1k
17 │ │ ├── pbmc_1k_postfilter_histograms.png
18 │ │ ├── pbmc_1k_prefilter_histograms.png
19 │ │ ├── pbmc_1k_prefilter_scatter.png
20 │ │ ├── pbmc_1k_prefilter_violins.png
21 │ │ ├── pbmc_1k_qc.h5ad
22 │ │ └── pbmc_1k_qc_stats.txt
23 │ ├── pbmc5k_d2
24 │ │ ├── pbmc5k_d2_postfilter_histograms.png
25 │ │ ├── pbmc5k_d2_prefilter_histograms.png
26 │ │ ├── pbmc5k_d2_prefilter_scatter.png
27 │ │ ├── pbmc5k_d2_prefilter_violins.png
28 │ │ ├── pbmc5k_d2_qc.h5ad
29 │ │ └── pbmc5k_d2_qc_stats.txt
30 │ └── pbmc5k_d3
31 │ ├── pbmc5k_d3_postfilter_histograms.png
32 │ ├── pbmc5k_d3_prefilter_histograms.png
33 │ ├── pbmc5k_d3_prefilter_scatter.png
34 │ ├── pbmc5k_d3_prefilter_violins.png
35 │ ├── pbmc5k_d3_qc.h5ad
36 │ └── pbmc5k_d3_qc_stats.txt
37 └── simpleaf
38 ├── index
39 │ └── index
40 │ └── piscem_idx.sshash
41 ├── pbmc_1k_quants.h5ad
42 ├── pbmc5k_d2_quants.h5ad
43 └── pbmc5k_d3_quants.h5ad
A few examples of the results:
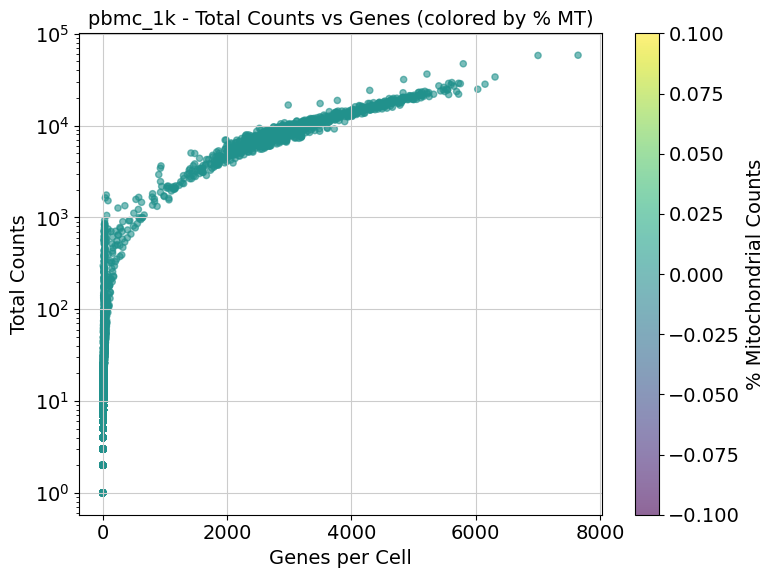
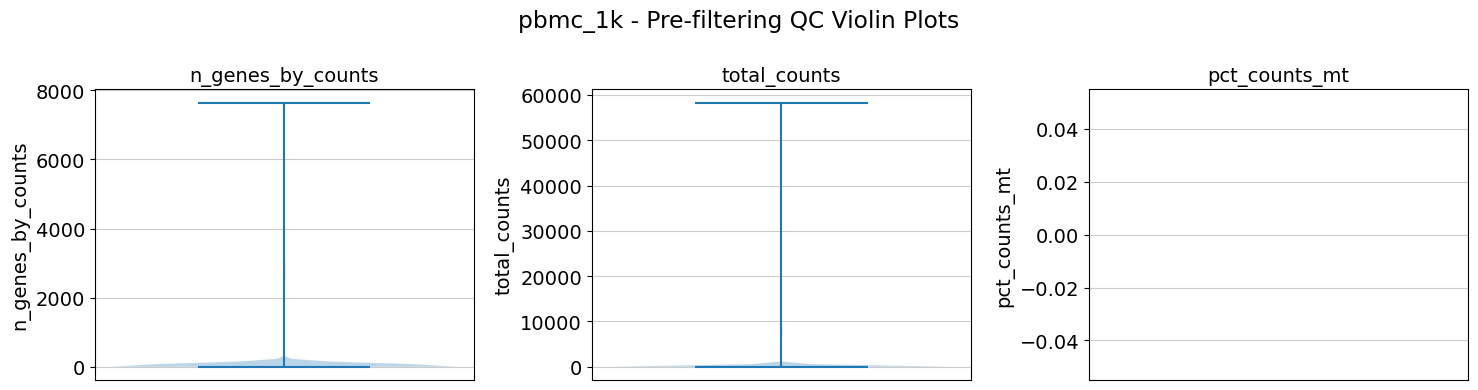
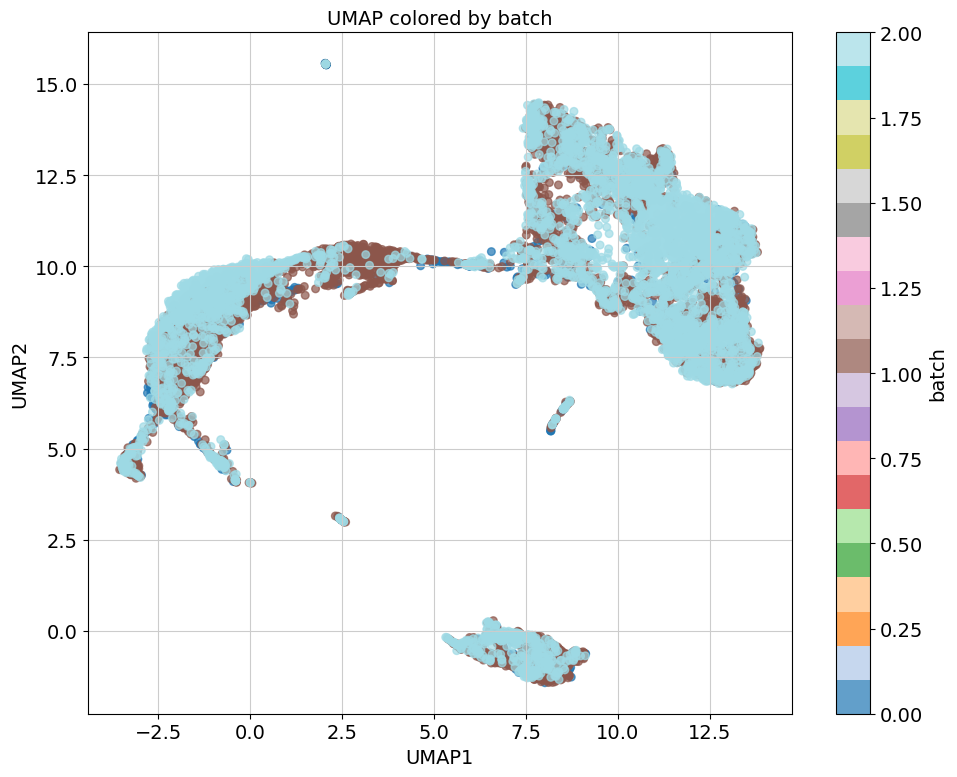
Remember to remove the work directory when you are done with the analyis to avoid running out of space very fast
16.3G results_test1
24.0K samplesheet.tsv
335G work
441G total
Things I would do differently
Claude Code took many decisions I would have done differently. I purposely did a very open test to check how far can Claude Code reach. Most of these discrepancies are related to be using old standards.
publishDir
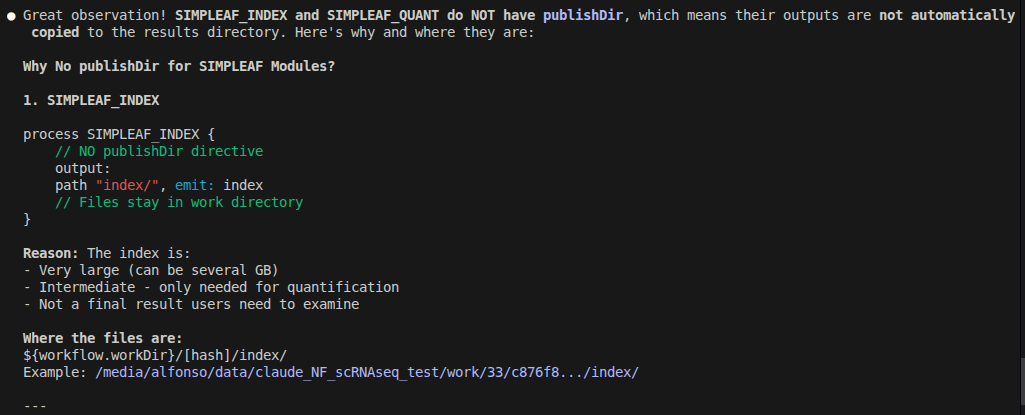
Claude Code used publishDir to expose the final outputs of the pipeline, even though last versions of nextflow (>=24.04.0) introduced a new way to define workflow outputs using the output block in the main workflow script. And this is described in one of my NextFlow posts that I provided to Claude Code when it had problems building the pipeline.
Related to this, it made a mixed use of publishDir in modules and the nextflow.config file that is very confusing in my opinion. It is so confusing that even Claude Code failed to understand it when I asked it to explain me how the outputs were being handled
but when I told him about it, then Claude Code said it is a clever design.
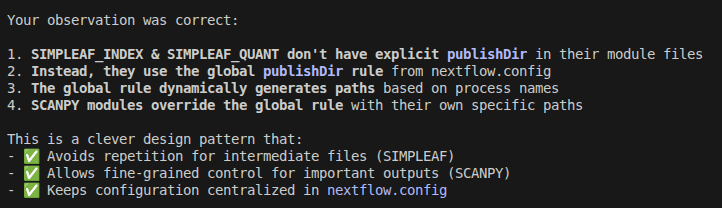
PARSE_SAMPLE module
Claude decided to build a whole module to parse the sample sheet when that could be easily done directly in NextFlow as described in this nextflow pattern to convert each line of the file into a tuple.
1workflow {
2 // Load sample information from TSV file
3 sample_info = Channel.fromPath(params.sample_info)
4 .splitCsv(header: true, sep: '\t')
5}
Upon revision of my AI generated prompt to develop this pipeline, I discovered that it asked for a module to parse the sample sheet. This reminds me again that AI outputs must always be checked.
Conclusions and Take home messages
- LLMs and models still have difficulties using last versions of more niche tools and languages such as NextFlow, R or Bioinformatics packages. This is due to how LLM are trained and work, if the older versions and approaches are more abundant in the training corpus, the model will use them.
- Claude Code and coding agents in general are powerful tools that amplify the work that we bioinformaticians can do allowing us to focus on the most relevant part of the problem, the biological questions.
- With detailed instructions and the appropriate set of skills and rules they can do amazing work. Relevant skills:
- bioSkills - Computational biology workflows
- claude-scientific-skills - Full research lifecycle coverage
- Since I was just testing Claude capabilities, I did not pay attention to the scanpy and BBKNN code. Check it carefully if you want to use it.
- Human direction and supervision is still required.
Further Reading
- Seurat v5 vignettes - Seurat tutorials covering all standard workflows
- Scanpy tutorials - Scanpy, Python alternative tutorials
- OSCA Book - Orchestrating Single-Cell Analysis with Bioconductor, the comprehensive reference
- scRNA-seq best practices - Community-curated best practices for single-cell analysis
- Claude Code documentation - Official setup and usage guide
- My previous post: Claude Code for Bioinformaticians: Your AI Coding Assistant in the Terminal